Introgressing the Aegilops tauschii genome into wheat as a basis for cereal improvement
Increasing crop production is necessary to feed the world’s expanding population, and crop breeders often utilize genetic variations to improve crop yield and quality. However, the narrow diversity of the wheat D genome seriously restricts its selective breeding. A practical solution is to exploit the genomic variations of Aegilops tauschii via introgression. Here, we established a rapid introgression platform for transferring the overall genetic variations of A. tauschii to elite wheats, thereby enriching the wheat germplasm pool. To accelerate the process, we assembled four new reference genomes, resequenced 278 accessions of A. tauschii and constructed the variation landscape of this wheat progenitor species. Genome comparisons highlighted diverse functional genes or novel haplotypes with potential applications in wheat improvement. We constructed the core germplasm of A. tauschii, including 85 accessions covering more than 99% of the species’ overall genetic variations. This was crossed with elite wheat cultivars to generate an A. tauschii-wheat synthetic octoploid wheat (A-WSOW) pool. Laboratory and field analysis with two examples of the introgression lines confirmed its great potential for wheat breeding. Our high-quality reference genomes, genomic variation landscape of A. tauschii and the A-WSOW pool provide valuable resources to facilitate gene discovery and breeding in wheat.(Nature Plant)
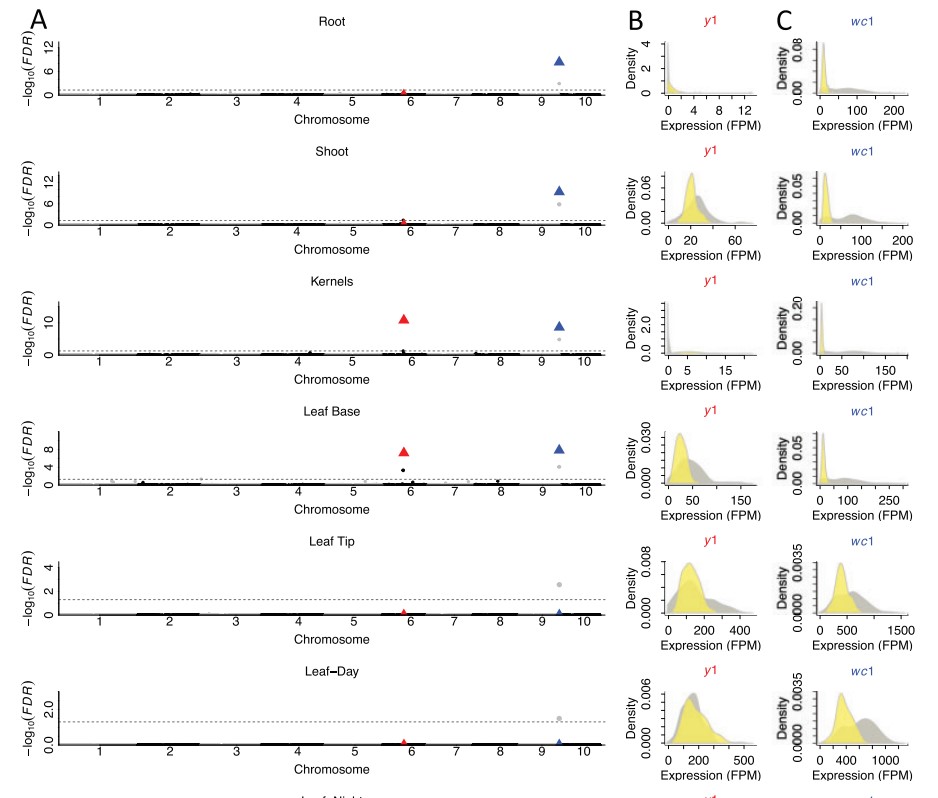
An inferred functional impact map of genetic variants in rice
Interpreting the functional impacts of genetic variants (GVs) is an important challenge for functional genomic studies in crops and next-generation breeding. Previous studies in rice (Oryza sativa) have focused mainly on the identification of GVs, whereas systematic functional annotation of GVs has not yet been performed. Here, we present a functional impact map of GVs in rice. We curated haplotype information for 17 397 026 GVs from sequencing data of 4726 rice accessions. We quantitatively evaluated the effects of missense mutations in coding regions in each haplotype based on the conservation of amino acid residues and obtained the effects of 918 848 non-redundant missense GVs. Furthermore, we generated high-quality chromatin accessibility (CA) data from six representative rice tissues and used these data to train deep convolutional neural network models to predict the impacts of 5 067 405 GVs for CA in regulatory regions. We characterized the functional properties and tissue specificity of the GV effects and found that large-effect GVs in coding and regulatory regions may be subject to selection in different directions. Finally, we demonstrated how the functional impact map could be used to prioritize causal variants in mapping populations. This impact map will be a useful resource for accelerating gene cloning and functional studies in rice, and can be freely queried in RiceVarMap V2.0 (http://ricevarmap.ncpgr.cn).(Molecular Plant)
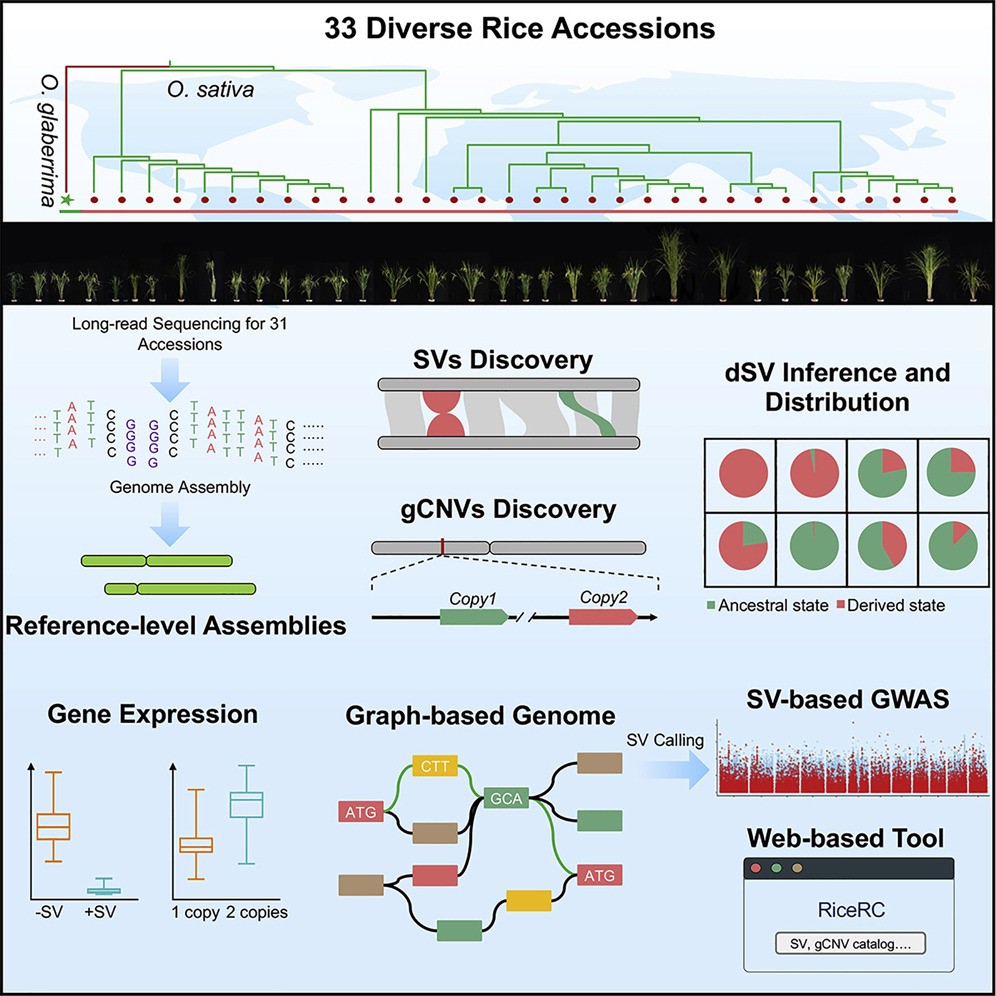
Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations
Structural variations (SVs) and gene copy number variations (gCNVs) have contributed to crop evolution, domestication, and improvement. Here, we assembled 31 high-quality genomes of genetically diverse rice accessions. Coupling with two existing assemblies, we developed pan-genome-scale genomic resources including a graph-based genome, providing access to rice genomic variations. Specifically, we discovered 171,072 SVs and 25,549 gCNVs and used an Oryza glaberrima assembly to infer the derived states of SVs in the Oryza sativa population. Our analyses of SV formation mechanisms, impacts on gene expression, and distributions among subpopulations illustrate the utility of these resources for understanding how SVs and gCNVs shaped rice environmental adaptation and domestication. Our graph-based genome enabled genome-wide association study (GWAS)-based identification of phenotype-associated genetic variations undetectable when using only SNPs and a single reference assembly. Our work provides rich population-scale resources paired with easy-to-access tools to facilitate rice breeding as well as plant functional genomics and evolutionary biology research.(Cell )
TWAS results are complementary to and less affected by linkage disequilibrium than GWAS
A genome-wide association study (GWAS) is used to identify genetic markers associated with phenotypic variation. In contrast, a transcriptome-wide association study (TWAS) detects associations between gene expression levels and phenotypic variation. It has previously been shown that in the cross-pollinated species, maize (Zea mays), GWAS, and TWAS identify complementary sets of trait-associated genes, many of which exhibit characteristics of true positives. Here, we extend this conclusion to the self-pollinated species, Arabidopsis thaliana and soybean (Glycine max). Linkage disequilibrium (LD) can result in the identification, via GWAS, of false-positive associations. In all three analyzed plant species, most trait-associated genes identified via TWAS are well separated physically from other candidate genes. Hence, TWAS is less affected by LD than is GWAS, demonstrating that TWAS is particularly well suited for association studies in genomes with slow rates of LD decay, such as soybean. TWAS is reasonably robust to the plant organs/tissues used to determine expression levels. In summary, this study confirms that TWAS is a promising approach for accurate gene-level association mapping in plants that is complementary to GWAS, and established that TWAS can exhibit substantial advantages relative to GWAS in species with slow rates of LD decay.(Plant Physiology)
A SNP based GWAS and functional haplotype based GWAS of fag leaf related traits and their infuence on the yield of bread wheat (Triticum aestivum L.)
The genetic architecture of five flag leaf morphology traits was dissected by the functional haplotype-based GWAS and a standard SNP-based GWAS in a diverse population consisting of 197 varieties. Abstract Flag leaf morphology (FLM) is a critical factor affecting plant architecture and grain yield in wheat. The genetic architecture of FLM traits has been extensively studied with QTL mapping in bi-parental populations, while few studies exploited genome-wide association studies (GW AS) in diverse populations. In this study, a panel of 197 elite and historical varieties from China was evaluated for five FLM traits including the length (FLL), width (FLW), ratio (FLR), area (FLA) and angle (FLANG) as well as yield in nine environments. Based on the phenotypic correlation between yield and FLL (-0.43), FLA (− 0.32) and FLW (0.11), an empirical FLM index combining the three FLM traits proved to be a good predictor for yield. Two GW AS approaches were applied to dissect the genetic architecture of five FLM traits with a Wheat660K SNP array. The functional haplotype-based GW AS revealed 6, 5 and 7 QTL for FLANG, FLL and FLR, respectively, whereas two QTL for FLW and one for FLR were identified by the standard SNP-based GW AS. Due to co-localization, there were 18 independent QTL and 10 of them were close to known ones. One co-localized QTL on chromosome 5A was associated with FLL, FLANG and FLR. Moreover, both GW AS approaches identified a novel QTL for FLR on chromosome 6B which was not reported in previous studies. This study provides new insights into the relationship between FLM and yield and broadens our understanding of the genetic architecture of FLM traits in wheat. (Theoretical and Applied Genetics)
Genome wide association mapping of leaf rust and stripe rust resistance in wheat accessions using the 90K SNP array
A genome-wide association analysis identified diverse loci for seedling and adult plant resistance to leaf rust and stripe rust. KASP markers were developed and validated for marker-assisted selection. Abstract Wheat leaf rust and stripe rust cause significant losses in many wheat producing regions worldwide. The objective of this study was to identify chromosome regions conferring resistance to both leaf rust and stripe rust at the seedling and adult plant stages. A diversity panel of 268 wheat lines, including 207 accessions from different wheat growing regions in China, and 61 accessions from foreign countries, were evaluated for leaf rust response at seedling stage using eight Chinese Puccinia triticina pathotypes, and also tested for leaf rust and stripe rust at adult plant stage in multiple field environments. The panel was genotyped with the Wheat 90 K Illumina iSelect SNP array. Genome-wide association mapping (GW AS) was performed using the mixed linear model (MLM). Twenty-two resistance loci including the known Lr genes, Lr1, Lr26, Lr3ka, LrZH22, and 18 potentially new loci were identified associated with seedling resistance, explaining 4.6 to 25.2% of the phenotypic variance. Twenty-two and 23 adult plant resistance (APR) QTL associated with leaf and stripe rust, respectively, were identified at adult stage, explaining 4.2–11.5% and 4.4–9.7% of the phenotypic variance. Among them, QLr-2BS was the potentially most valuable all-stage resistance gene. Seven and six consistent APR QTL were identified in multiple environments including best linear unbiased prediction (BLUP) data, respectively. Comparison with previously mapped resistance loci indicated that three of the seven leaf rust resistance APR QTL, and two of the six stripe rust resistance APR QTL were new. Four potentially pleiotropic APR QTL, including Lr46/Yr29, QLr-2AL.1/QYr-2AL.1, QLr-2AL.2/QYr-2AL.2, and QLr-5BL/QYr-5BL.1, were identified. Twelve associated SNPs were converted into kompetitive allele-specific PCR (KASP) markers and verified in bi-parental populations. The study reports genetic loci conferring resistance to both diseases, and the closely linked markers should be applicable for marker-assisted wheat breeding.(Theoretical and Applied Genetics)
Cytological and molecular characteristics of delayed spike development in wheat under low temperature in early spring
Low temperature in early spring impairs wheat growth and grain yield. However, little is known about the cytological and molecular mechanisms underlying low temperature regulation of wheat spike development. Microstructure observation and transcriptome sequencing of wheat spikes under low temperature were conducted. Low temperature slowed spike development, reduced the yield-component parameters of wheat spikes at the harvest stage, delayed the formation of lateral spikelet and tissue development, and induced the early differentiation of terminal spirelets. Low temperature increased the content of abscisic acid and caused the upregulation of genes in the abscisic acid signaling pathway, including those encoding PP2Cs, SnRK2s, and bZIP transcription factors. Low temperature also induced the upregulation of 33 cold-responsive genes involved in wheat response to low-temperature stress and regulation of abscisic acid biosynthesis and metabolism of other substances. The wheat spike adapted to cold conditions by changing the expression levels of genes involved in spike morphogenesis, including the transcription-factor genes MADS6, ERF4, ERF78, WOX6, and NAC48. These findings suggest that low temperature in early spring delays wheat spike development by increasing abscisic acid content or affecting the expression of genes involved in morphogenesis. (The Crop Journal)

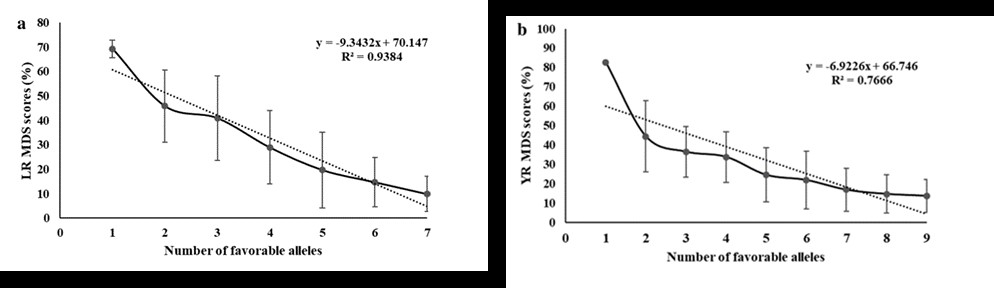
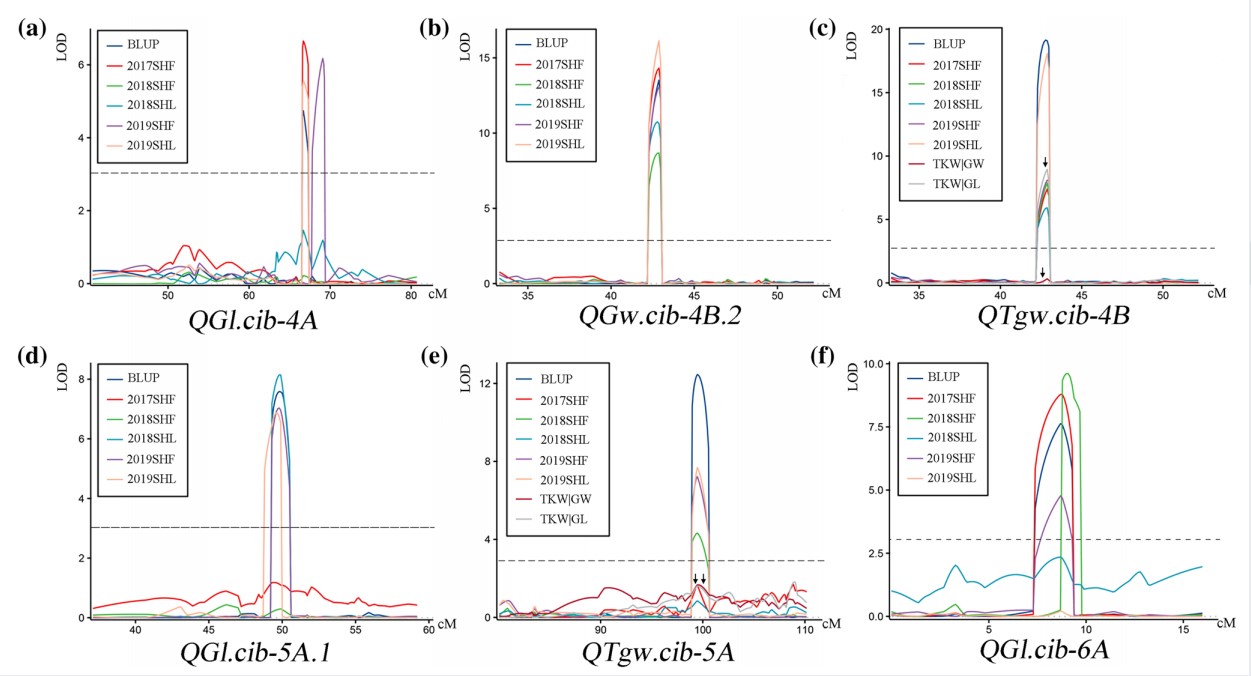
Genetic dissection of quantitative trait loci for grain size and weight by high-resolution genetic mapping in bread wheat (Triticum aestivum L.)
Grain size and weight are crucial components of wheat yield. Dissection of their genetic control is thus essential for the improvement of yield potential in wheat breeding. We used a doubled haploid (DH) population to detect quantitative trait loci (QTLs) for grain width (GW), grain length (GL), and thousand grain weight (TGW) in fve environments. Six major QTLs, QGw.cib-4B.2, QGl.cib-4A, QGl.cib-5A.1, QGl.cib-6A, QTgw.cib-4B, and QTgw.cib-5A, were consistently identifed in at least three individual environments and in best linear unbiased prediction (BLUP) datasets, and explained 5.65–34.06% of phenotypic variation. QGw.cib-4B.2, QTgw.cib-4B, QGl.cib-5A.1 and QGl.cib-6A had no efect on grain number per spike (GNS). In addition to QGl.cib-4A, the other major QTLs were further validated by using Kompetitive Allele Specifc PCR (KASP) markers in diferent genetic backgrounds. Moreover, signifcant interactions between the three major GL QTLs and two major TGW QTLs were observed. Comparison analysis showed that QGl.cib-5A.1 and QGl.cib-6A are likely new loci. Notably, QGw.cib-4B.2 and QTgw.cib-4B were co-located on chromosome 4B and improved TGW by increasing only GW, unlike nearby or overlapped loci reported previously. Three genes associated with grain development within the QGw.cib-4B.2/QTgw.cib-4B interval were identifed by searches on sequence similarity, spatial expression patterns, and orthologs. The major QTLs and KASP markers reported here will be useful for elucidating the genetic architecture of grain size and weight and for developing new wheat cultivars with high and stable yield.(Theoretical and Applied Genetics)
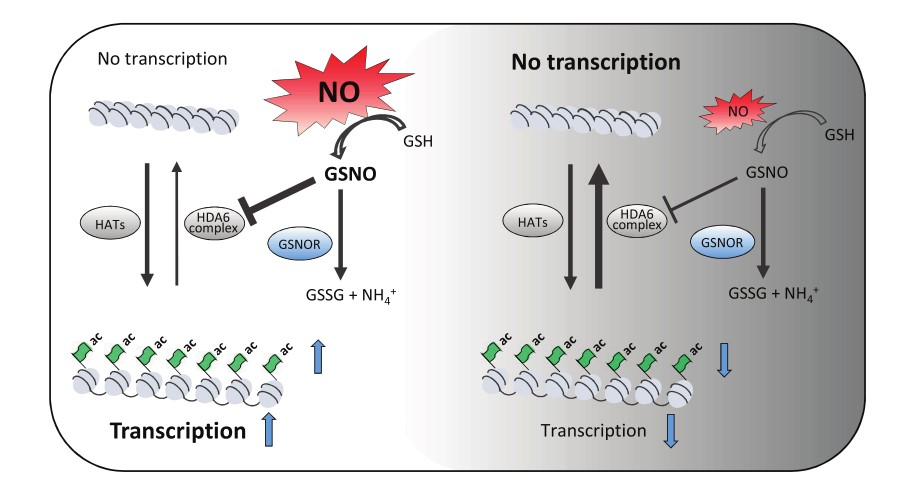
Nitric oxide coordinates growth, development, and stress response via histone modification and gene expression
Nitric oxide (NO) is a signaling molecule with multiple regulatory functions in plant physiology and stress response. In ad-dition to direct effects on transcriptional machinery, NO executes its signaling function via epigenetic mechanisms. We re-port that light intensity-dependent changes in NO correspond to changes in global histone acetylation (H3, H3K9, andH3K9/K14) in Arabidopsis (Arabidopsis thaliana) wild-type leaves, and that this relationship depends on S-nitrosogluta-thione reductase and histone deacetylase 6 (HDA6). The activity of HDA6 was sensitive to NO, demonstrating that NOparticipates in regulation of histone acetylation. Chromatin immunoprecipitation sequencing and RNA-seq analysesrevealed that NO participates in the metabolic switch from growth and development to stress response. This coordinating function of NO might be particularly important in plant ability to adapt to a changing environment, and is therefore a promising foundation for mitigating the negative effects of climate change on plant productivity.(Plant Physiology)
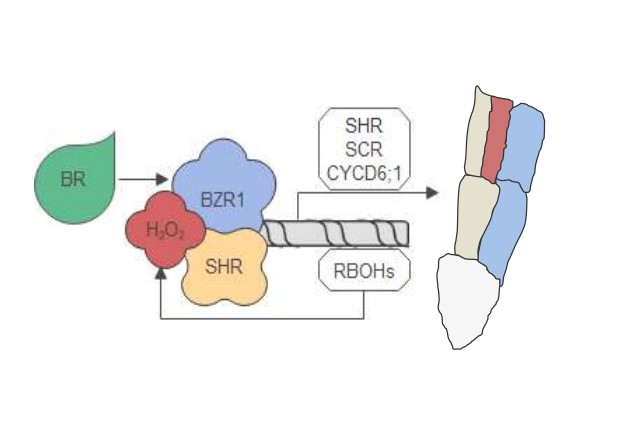
Integrated regulation of periclinal cell division by transcriptional module of BZR1-SHR in Arabidopsis roots
● The timing and extent of cell division are crucial for the correct patterning of multicellularorganism. In Arabidopsis, root ground tissue maturation involves the periclinal cell division of the endodermis to generate two cell layers: endodermis and middle cortex. However, the molecular mechanism underlying this pattern formation remains unclear. ● Here, we report that phytohormone brassinosteroid (BR) and redox signal hydrogen peroxide (H2O2) interdependently promote periclinal division during root ground tissue maturation by regulating the activity of SHORT-ROOT (SHR), a master regulator of root growth and development. ● BR-activated transcription factor BRASSINAZOLE RESISTANT1 (BZR1) directly binds to the promoter of SHR to induce its expression, and physically interacts with SHR to increase the transcripts of RESPIRATORY BURST OXIDASE HOMOLOGs (RBOHs) and elevate the levels of H2O2, which feedback enhances the interaction between BZR1 and SHR. Additionally, genetic analysis shows that SHR is required for BZR1-promoted periclinal division, and BZR1 enhances the promoting effects of SHR on periclinal division. ● Together, our finding reveals that the transcriptional module of BZR1-SHR fine-tunes periclinal division during root ground tissue maturation in response to hormone and redox signals.(New Phytologist)
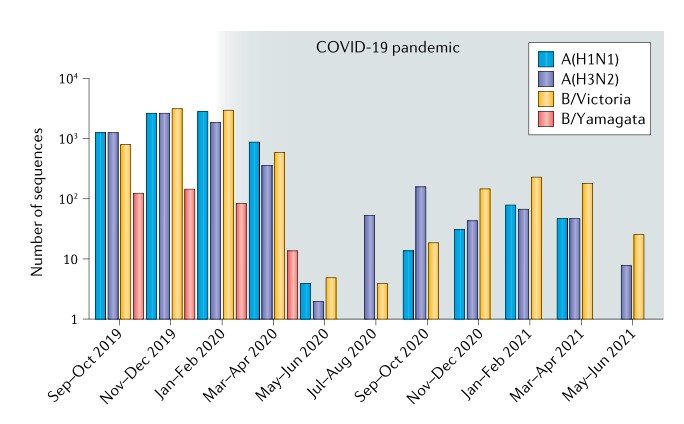
Influenza lineage extinction during the COVID-19 pandemic?
The SARS-CoV-2 pandemic has seen a notable global reduction in influenza cases of both influenza A and B viruses. In particular, the B/Yamagata lineage has not been isolated from April 2020 to August 2021, suggesting that this influenza lineage may have become extinct, which may provide opportunities for improving availability and effectiveness of influenza vaccines.(Nature Reviews Microbiology)
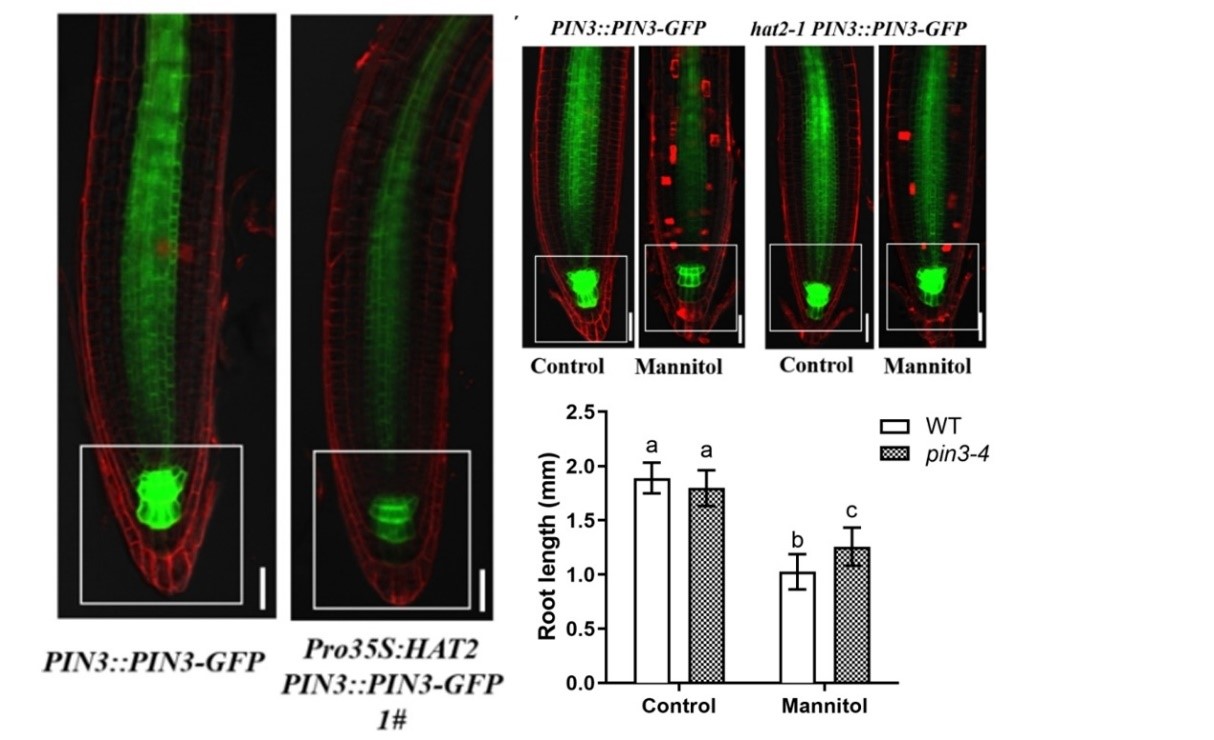
Osmotic stress represses root growth by modulating the transcriptional regulation of PIN-FORMED3
• Osmotic stress influences root system architecture, and polar auxin transport (PAT) is well-established to regulate root growth and development. However, how PAT responds to osmotic stress at the molecular level remains poorly understood. In this study, we explored whether and how the auxin efflux carrier PIN-FORMED3 (PIN3) participates in osmotic stress-induced root growth inhibition in Arabidopsis (Arabidopsis thaliana). • We observed that osmotic stress induces a HD-ZIP II transcription factor-encoding gene HOMEODOMAIN ARABIDOPSIS THALIANA2 (HAT2) expression in roots. The hat2 loss-of-function mutant is less sensitive to osmotic stress in terms of root meristem growth. Consistent with this phenotype, whereas the auxin response is down-regulated in wild-type roots under osmotic stress, the inhibition of auxin response by osmotic stress was alleviated in hat2 roots. Conversely, transgenic lines overexpressing HAT2 (Pro35S::HAT2) had shorter roots and reduced auxin accumulation compared with wild-type plants. • PIN3 expression was significantly reduced in the Pro35S::HAT2 lines. We determined that osmotic stress-mediated repression of PIN3 was alleviated in the hat2 mutant because HAT2 normally binds to the promoter of PIN3 and inhibits its expression. • Taken together, our data reveal that osmotic stress inhibits root growth via HAT2, which regulates auxin activity by directly repressing PIN3 transcription.(New Phytologist )
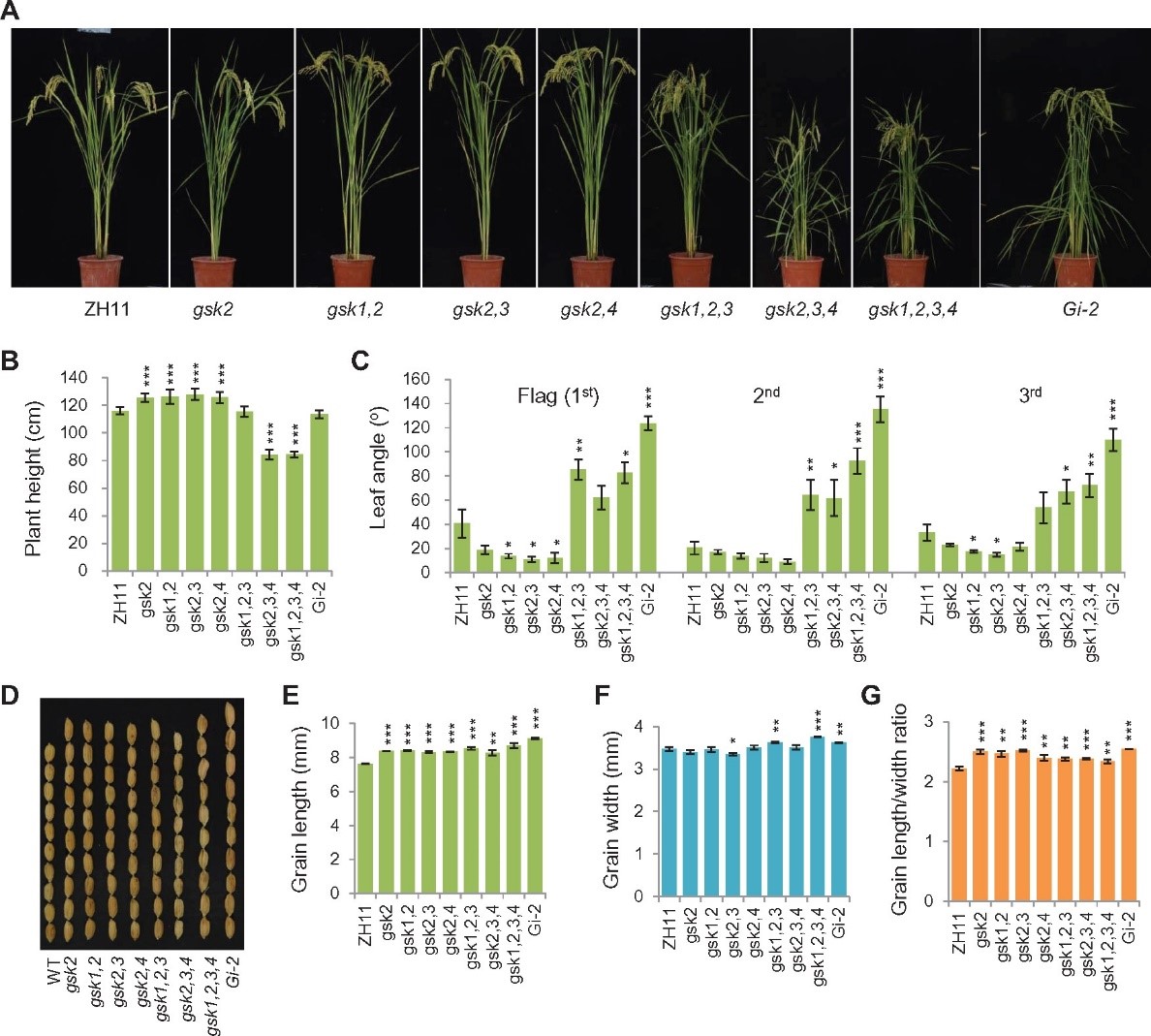
Diversification of plant agronomic traits by genome editing of brassinosteroid signaling family genes in rice
Brassinosteroids (BRs) regulate various agronomic traits such as plant height, leaf angle, and grain size in rice (Oryza sativaL.); thus, BR signaling components are promising targets for molecular rational design. However, genetic materials for BR-signaling genes or family members remain limited in rice. Here, by genome editing using clustered regularly interspaced short palindromic repeats (CRSPR)/Cas9 tools, we generated a panel of single, double, triple, or quadruple mutants within three BR signaling gene families, including GSK3/SHAGGY-LIKE KINASE1 (GSK1)–GSK4, BRASSINAZOLE-RESISTANT1(OsBZR1)–OsBZR4, and protein phosphatases with kelch-like (PPKL)1–PPKL3, under the same background (Zhonghua11, japonica). The high-order mutants were produced by either simultaneously targeting multiple sites on different genes of one family (GSKs and PPKLs) or targeting the overlapping sequences of family members (OsBZRs). The mutants exhibited a diversity of plant height, leaf angle, and grain morphology. Comparison analysis of the phenotypes together with BR sensitivity tests suggested the existence of functional redundancy, differentiation, or dominancy among the members within each family. In addition, we generated a set of transgenic plants overexpressing GSK2, OsBZR1/2, and PPKL2, respectively, in wild-type or activated forms with fusion of different tags, and also verified the protein response to BR application. Collectively, these plants greatly enriched the diversity of important agronomic traits in rice. We propose that editing of BR-related family genes could be a feasible approach for screening of desired plants to meet different requirements. Releaseof these materials as well as the related information also provides valuable resources for further BR research and utilization.(Plant Physiology)
De novo biosynthesis of bioactive isoflavonoids by engineered yeast cell factories
Isoflavonoids comprise a class of plant natural products with great nutraceutical, pharmaceutical and agricultural significance. Their low abundance in nature and structural complexity however hampers access to these phytochemicals through traditional crop-based manufacturing or chemical synthesis. Microbial bioproduction therefore represents an attractive alternative. Here, we engineer the metabolism of Saccharomyces cerevisiae to become a platform for efficient production of daidzein, a core chemical scaffold for isoflavonoid biosynthesis, and demonstrate its application towards producing bioactive glucosides from glucose, following the screening-reconstruction-application engineering framework. First, we rebuild daidzein biosynthesis in yeast and its production is then improved by 94-fold through screening biosynthetic enzymes, identifying rate-limiting steps, implementing dynamic control, engineering substrate trafficking and fine-tuning competing metabolic processes. The optimized strain produces up to 85.4 mg L−1 of daidzein and introducing plant glycosyltransferases in this strain results in production of bioactive puerarin (72.8 mg L−1) and daidzin (73.2 mg L−1). Our work provides a promising step towards developing synthetic yeast cell factories for de novo biosynthesis of value-added isoflavonoids and the multi-phased framework may be extended to engineer pathways of complex natural products in other microbial hosts. (Nature Communications)
A rice gene that confers broad-spectrum resistance to b-triketone herbicides
The genetic variation of rice cultivars provides a resource for further varietal improvement through breeding. Some rice varieties are sensitive to benzobicyclon (BBC), a β-triketone herbicide that inhibits 4-hydroxyphenylpyruvate dioxygenase (HPPD). Here we identify a rice gene, HIS1 (HPPD INHIBITOR SENSITIVE 1), that confers resistance to BBC and other β-triketone herbicides. We show that HIS1 encodes an Fe(II)/2-oxoglutarate-dependent oxygenase that detoxifies β-triketone herbicides by catalyzing their hydroxylation. Genealogy analysis revealed that BBC-sensitive rice variants inherited a dysfunctional his1 allele from an indica rice variety. Forced expression of HIS1 in Arabidopsis conferred resistance not only to BBC but also to four additional β-triketone herbicides. HIS1 may prove useful for breeding herbicide-resistant crops.(Science)
Characterization of the Durum Wheat-Aegilops tauschii 4D(4B) Disomic Substitution Line YL-443 With Superior Characteristics of High Yielding and Stripe Rust Resistance
Durum wheat is one of the important food and cash crops. The main goals in current breeding programs are improving its low yield potential, kernel characteristics, and lack of resistance or tolerance to some biotic and abiotic stresses. In this study, a nascent synthesized hexaploid wheat Lanmai/AT23 is used as the female parent in crosses with its AB genome donor Lanmai. A tetraploid line YL-443 with supernumerary spikelets and high resistance to stripe rust was selected out from the pentaploid F7 progeny. Somatic analysis using multicolor fluorescence in situ hybridization (mc-FISH) revealed that this line is a disomic substitution line with the 4B chromosome pair of Lanmai replaced by the 4D chromosome pair of Aegilops tauschii AT23. Comparing with Lanmai, YL-443 shows an increase in the number of spikelets and florets per spike by 36.3 and 75.9%, respectively. The stripe rust resistance gene Yr28 carried on the 4D chromosome was fully expressed in the tetraploid background. The present 4D(4B) disomic substitution line YL-443 was distinguished from the previously reported 4D(4B) lines with the 4D chromosomes from Chinese Spring (CS). Our study demonstrated that YL-443 can be used as elite germplasm for durum wheat breeding targeting high yield potential and stripe rust resistance. The Yr28-specific PCR marker and the 4D chromosome-specific KASP markers together with its unique features of pubescent leaf sheath and auricles can be utilized for assisting selection in breeding.(Front. Plant Sci. )

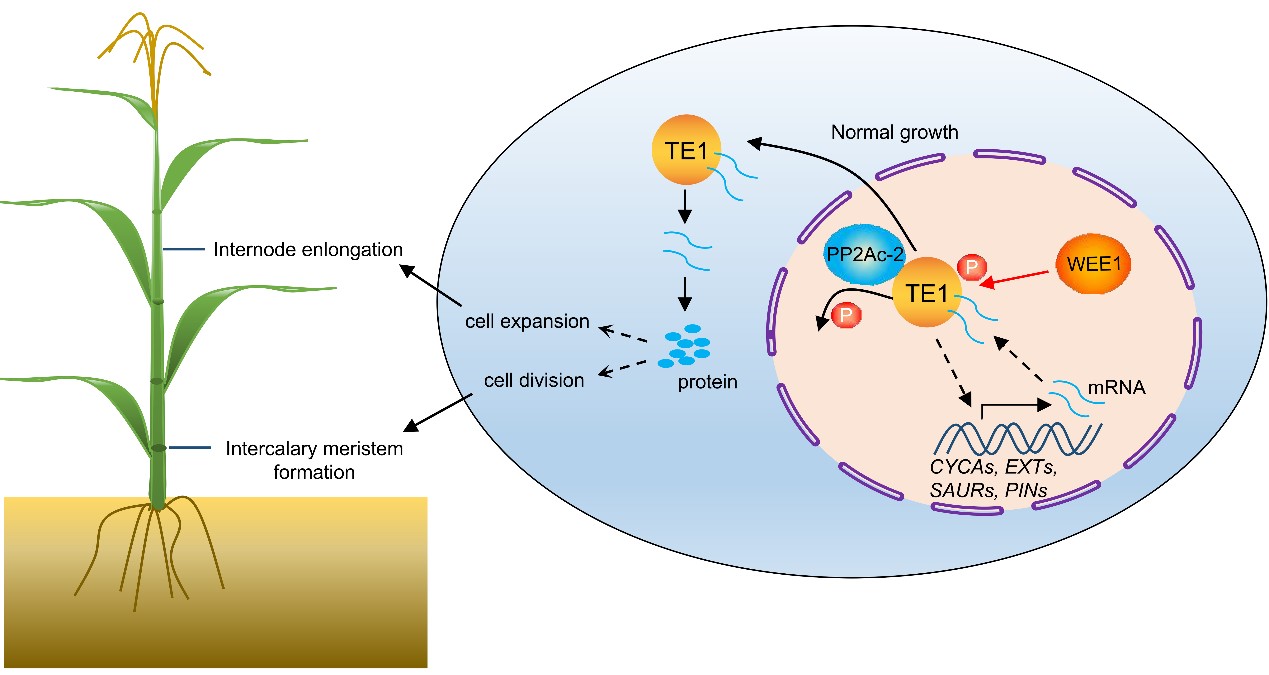
ZmTE1 promotes plant height by regulating intercalary meristem formation and internode cell elongation in maize
Maize height is determined by the number of nodes and the length of internodes. Node number is driven by intercalary meristem formation and internode length by intercalary cell elongation respectively. However, mechanisms regulating establishment of nodes and internode growth is unclear. We screened EMS-induced maize mutants and identified a dwarf mutant zm66, linked to a single base change in TERMINAL EAR 1 (ZmTE1). Detailed phenotypic analysis revealed that zm66 (zmte1-2) has shorter internodes and increased node numbers, caused by decreased cell elongation and disordered intercalary meristem formation, respectively. Transcriptome analysis showed that auxin signaling genes are also dysregulated in zmte1-2, as are cell elongation and cell cycle-related genes. This argues that ZmTE1 regulates auxin signaling, cell division and cell elongation. We found that the ZmWEE1 kinase phosphorylates ZmTE1, thus confining it to the nucleus and probably reducing cell division. In contrast, the ZmPP2Ac-2 phosphatase promotes dephosphorylation and cytoplasmic localization of ZmTE1, as well as cell division. Taken together, ZmTE1, a key regulator of plant height, is responsible for maintaining organized formation of internode meristems and rapid cell elongation. ZmWEE1 and ZmPP2Ac-2 might balance ZmTE1 activity, controlling cell division and elongation to maintain normal maize growth.(Plant Biotechnol J.)