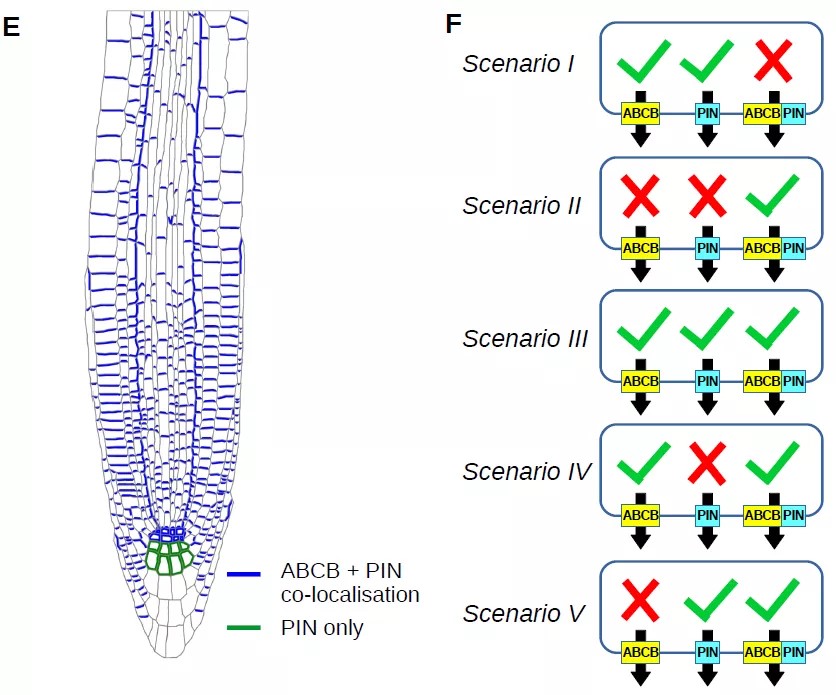
Systems approaches reveal that ABCB and PIN proteins mediate co-dependent auxin efflux
Members of the B family of membrane-bound ATP-binding cassette (ABC) transporters represent key components of the auxin-efflux machinery in plants. Over the last two decades experimental studies have shown that modifying ABCB expression affects auxin distribution and plant phenotypes. However, precisely how ABCB proteins transport auxin in conjunction with the more widely studied family of PIN auxin efflux transporters is unclear, and studies using heterologous systems have produced conflicting results. Here, we integrate ABCB localization data into a multicellular model of auxin transport in the Arabidopsis root tip to predict how ABCB-mediated auxin transport impacts organ-scale auxin distribution. We use our model to test five potential ABCB-PIN regulatory interactions, simulating the auxin dynamics for each interaction and quantitatively comparing the predictions with experimental images of the DII-VENUS auxin reporter in wild type and abcb single and double loss-of-function mutants. Only specific ABCB-PIN regulatory interactions result in predictions that recreate the experimentally observed DII-VENUS distributions and long-distance auxin transport. Our results suggest that ABCBs enable auxin efflux independently of PINs; however, PIN-mediated auxin efflux is predominantly through a co- dependent efflux where co-localised with ABCBs.(Plant Cell)
Stress granule-associated TaMBF1c confers thermotolerance through regulating specific mRNA translation in wheat (Triticum aestivum)
Heat stress is a major limiting factor for global wheat production and causes dramatic yield loss worldwide. The TaMBF1c gene is upregulated in response to heat stress in wheat. Understanding the molecular mechanisms associated with heat stress responses will pave the way to improve wheat thermotolerance. Through CRISPR/Cas9-based gene editing, polysome profiling coupled with RNA-sequencing analysis, and protein–protein interactions, we show that TaMBF1c conferred heat response via regulating a specific gene translation in wheat. The results showed that TaMBF1c is evolutionarily conserved in diploid, tetraploid and hexaploid wheat species, and its knockdown and knockout lines show increased heat sensitivity. TaMBF1c is colocalized with the stress granule complex and interacts with TaG3BP.TaMBF1c affects the translation efficiency of a subset of heat responsive genes, which are significantly enriched in the ‘sequence-specific DNA binding’ term. Moreover, gene expression network analysis demonstrated that TaMBF1c is closely associated with the translation of heat shock proteins.Our findings reveal a contribution of TaMBF1c in regulating the heat stress response via the translation process, and provide a new target for improving heat tolerance in wheat breeding programs.(New phytologist)
UDP-N-acetylglucosamine pyrophosphorylase enhances rice survival at high temperature
High-temperature stress inhibits normal cellular processes and results in abnormal growth and development in plants. However, the mechanisms by which rice (Oryza sativa) copes with high temperature are not yet fully understood. In this study, we identified a rice high temperature enhanced lesion spots 1 (hes1) mutant, which displayed larger and more dense necrotic spots under high temperature conditions. HES1 encoded a UDP-N-acetylglucosamine pyrophosphorylase, which had UGPase enzymatic activity. RNA sequencing analysis showed that photosystem-related genes were differentially expressed in the hes1 mutant at different temperatures, indicating that HES1 plays essential roles in maintaining chloroplast function. HES1 expression was induced under high temperature conditions. Furthermore, loss-of-function of HES1 affected heat shock factor expression and its mutation exhibited greater vulnerability to high temperature. Several experiments revealed that higher accumulation of reactive oxygen species occurred in the hes1 mutant at high temperature. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and comet experiments indicated that the hes1 underwent more severe DNA damage at high temperature. The determination of chlorophyll content and chloroplast ultrastructure showed that more severe photosystem defects occurred in the hes1 mutant under high temperature conditions. This study reveals that HES1 plays a key role in adaptation to high-temperature stress in rice.(New phytologist)
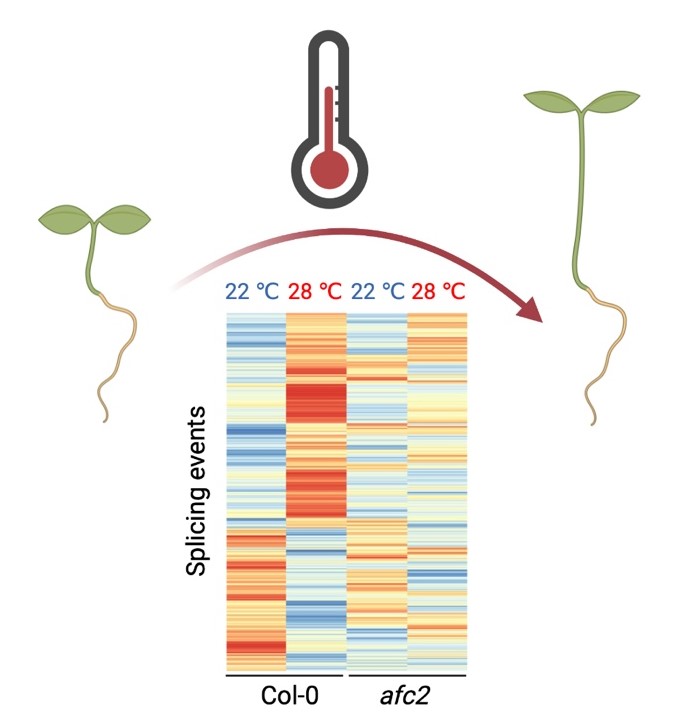
Plant AFC2 kinase desensitizes thermomorphogenesis through modulation of alternative splicing
High ambient temperatures have adverse impacts on crop yields. Although a few plant thermosensors have been reported, these sensors directly or indirectly impact PIF4-controlled transcriptional regulation. Moreover, high temperatures also trigger a number of post-transcriptional alternative splicing events in plants and even in animals. Here, we show that LAMMER kinase AFC2 in Arabidopsis controls high-temperature-triggered alternative splicing. Plants without AFC2 exhibited distorted splicing patterns at a high ambient temperature. Further investigations revealed that high temperatures triggered alternative splicing in the majority of PIF4 target genes as a means of desensitizing PIF4 signaling. Consistently, the afc2 mutants exhibited more exaggerated high ambient temperature responses in a PIF4-dependent manner. AFC2 directly phosphorylated the serine/arginine-rich protein splicing factor RSZ21, and AFC2 kinase activity decreased with increasing temperature, indicating that the AFC2 itself may sense temperature changes. In summary, we report that alternative splicing is a safe-guard mechanism when plants encounter high temperature.(iScience)
TT2 controls rice thermotolerance through SCT1-dependent alteration of wax biosynthesis
Global warming threatens crop production. G proteins mediate plant responses to multiple abiotic stresses. Here we identified a natural quantitative trait locus, TT2 (THEROMOTOLERANCE 2), encoding a Gγ subunit, that confers thermotolerance in rice during both vegetative and reproductive growth without a yield penalty. A natural allele with loss of TT2 function was associated with greater retention of wax at high temperatures and increased thermotolerance. Mechanistically, we found that a transcription factor, SCT1 (Sensing Ca2+ Transcription factor 1), functions to decode Ca2+ through Ca2+-enhanced interaction with calmodulin and acts as a negative regulator of its target genes (for example, Wax Synthesis Regulatory 2 (OsWR2)). The calmodulin-SCT1 interaction was attenuated by reduced heat-triggered Ca2+ caused by disrupted TT2, thus explaining the observed heat-induced changes in wax content. Beyond establishing a bridge linking G protein, Ca2+ sensing and wax metabolism, our study illustrates innovative approaches for developing potentially yield-penalty-free thermotolerant crop varieties.(Nature Plants)
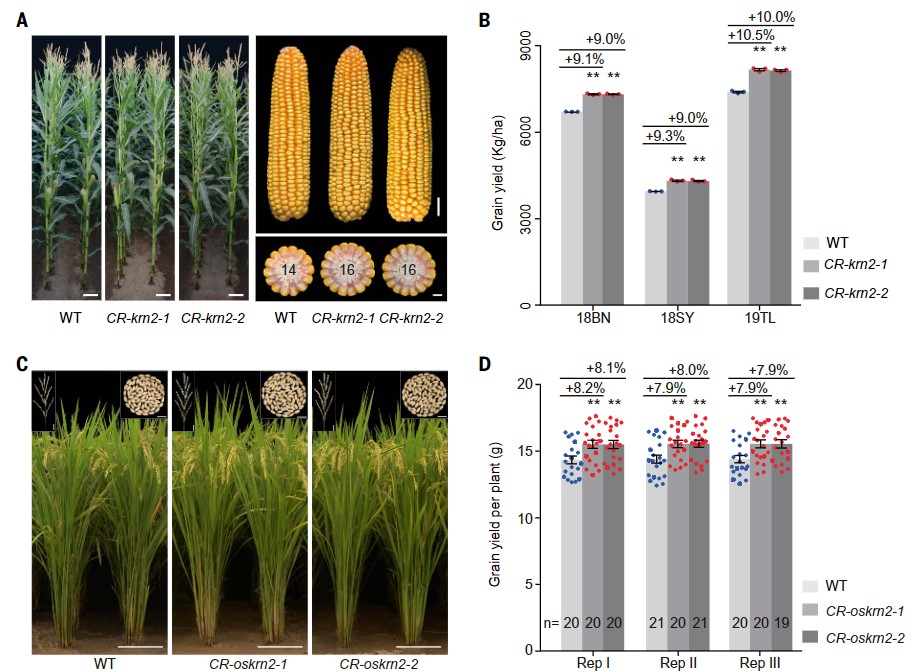
Convergent selection of a WD40 protein that enhances grain yield in maize and rice
A better understanding of the extent of convergent selection among crops could greatly improve breeding programs. We found that the quantitative trait locus KRN2 in maize and its rice ortholog, OsKRN2, experienced convergent selection. These orthologs encode WD40 proteins and interact with a gene of unknown function, DUF1644, to negatively regulate grain number in both crops. Knockout of KRN2 in maize or OsKRN2 in rice increased grain yield by ~10% and ~8%, respectively, with no apparent trade-offs in other agronomic traits. Furthermore, genome-wide scans identified 490 pairs of orthologous genes that underwent convergent selection during maize and rice evolution, and these were enriched for two shared molecular pathways. KRN2, together with other convergently selected genes, provides an excellent target for future crop improvement.(Science)
Multi-omics analysis dissects the genetic architecture of seed coat content in Brassica napus
Here we provide insights into the genetic basis of natural variation of seed coat content by transcriptome-wide association studies (TWAS) and genome-wide association studies (GWAS) using 382 B. napus accessions. By population transcriptomic analysis, we identify more than 700 genes and four gene modules that are significantly associated with seed coat content. We also characterize three reliable quantitative trait loci (QTLs) controlling seed coat content by GWAS. Combining TWAS and correlation networks of seed coat content-related gene modules, we find that BnaC07.CCR-LIKE (CCRL) and BnaTT8s play key roles in the determination of the trait by modulating lignin biosynthesis. By expression GWAS analysis, we identify a regulatory hotspot on chromosome A09, which is involved in controlling seed coat content through BnaC07.CCRL and BnaTT8s. We then predict the downstream genes regulated by BnaTT8s using multi-omics datasets. We further experimentally validate that BnaCCRL and BnaTT8 positively regulate seed coat content and lignin content. BnaCCRL represents a novel identified gene involved in seed coat development. Furthermore, we also predict the key genes regulating carbon allocation between phenylpropane compounds and oil during seed development in B. napus.(Genome Biology)
Rice OsIAA6 interacts with OsARF1 and regulates leaf inclination
Leaf inclination, a component of crop architecture, influences photosynthetic efficiency and planting density. Various factors, particularly the phytohormones auxin and brassinosteroids (BRs), function in regulating lamina joint bending, and understanding of the genetic control of leaf inclination will help to elucidate the relevant regulatory network. Screening a rice T-DNA insertion population revealed a mutant that was insensitive to auxin and displayed an enlarged leaf angle due to increased cell length on the adaxial side of the lamina joint. Genetic analysis revealed that the increased leaf inclination was caused by T-DNA insertion in the promoter region of OsIAA6, resulting in elevated OsIAA6 expression. Further study showed that OsIAA6 interacts with OsARF1 to suppress auxin signaling and regulates leaf inclination. OsIAA6 mediates the BR effects on lamina joint development, and OsBZR1, the key transcription factor in BR signaling, binds directly to the promoter of OsIAA6 to stimulate its transcription. These results indicate the roles of the OsIAA6–OsARF1 module in regulating rice leaf inclination and suggest the synergistic effects of the phytohormones auxin and BR. (The Crop Journal)
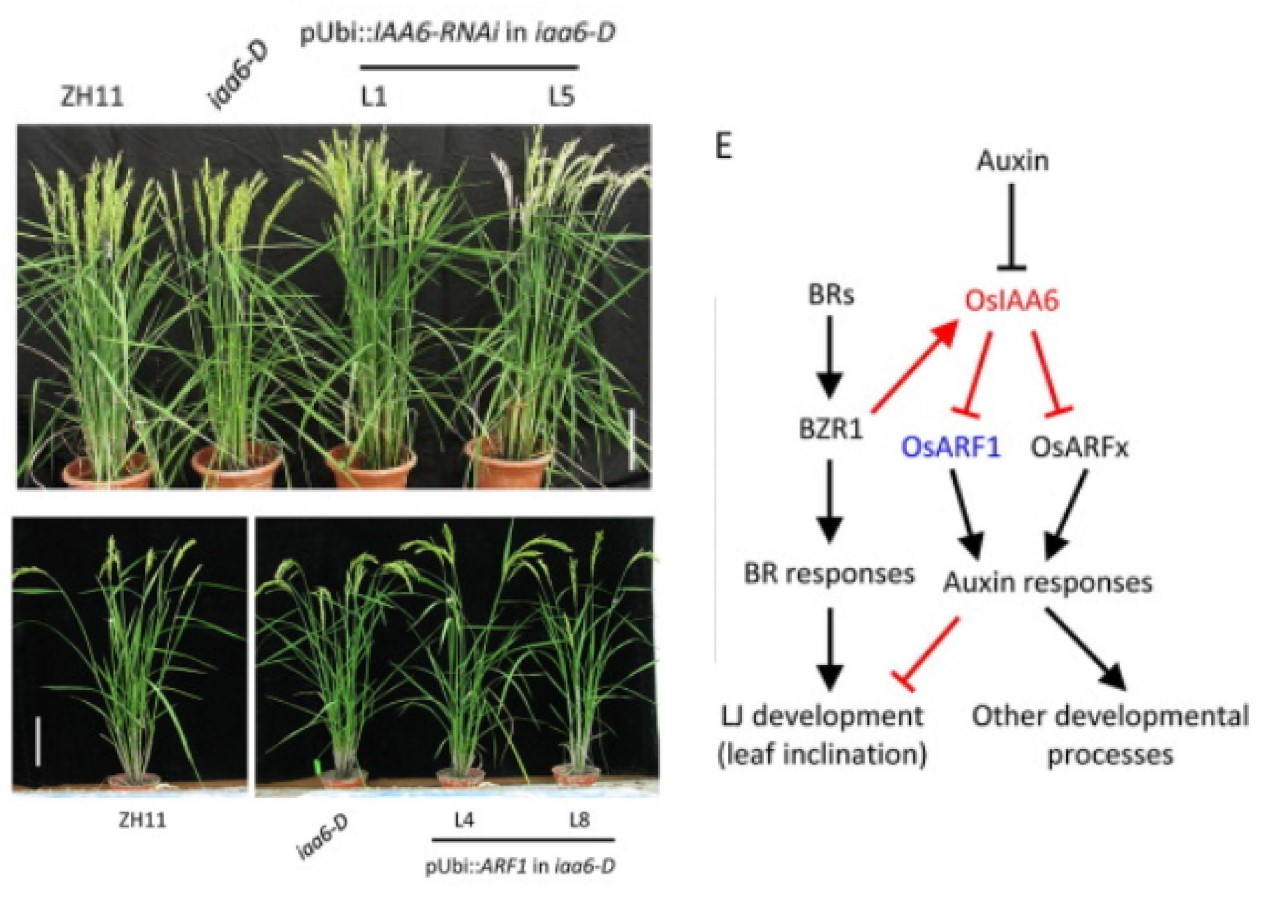
OsSLA1 functions in leaf angle regulation by enhancing the interaction between OsBRI1 and OsBAK1 in rice
Leaf angle is an important trait in plants. Here, we demonstrate that the leucine-rich-repeat receptor-like kinase (LRR-RLK) OsSLA1 plays an important role in leaf angle regulation. OsSLA1 mutant plants exhibited a small leaf angle phenotype due to changes of adaxial cells in the lamina joint. GUS staining revealed that OsSLA1 was highly expressed in adaxial cells of the lamina joint. The OsSLA1 mutant plants were insensitive to exogenous eBL and showed up-regulated expression of DWARF and CPD, but down-regulated expression of BU1, BUL1, and ILI1, indicating that BR signal transduction was blocked. Fluorescence microscopy showed that OsSLA1 was localized to the plasma membrane and nearby periplasmic vesicles. Further study showed that OsSLA1 interacts with OsBRI1 and OsBAK1 via its intracellular domain and promotes the interaction between OsBRI1 and OsBAK1. In addition, phosphorylation experiments revealed that OsSLA1 does not possess kinase activity, but that it can be phosphorylated by OsBRI1 in vitro. The knockout of OsSLA1 in the context of d61 caused exacerbation of the mutant phenotype. These results demonstrate that OsSLA1 regulates leaf angle formation via positive regulation of BR signaling by enhancing the interaction of OsBRI1 with OsBAK1.(The Plant Journal)

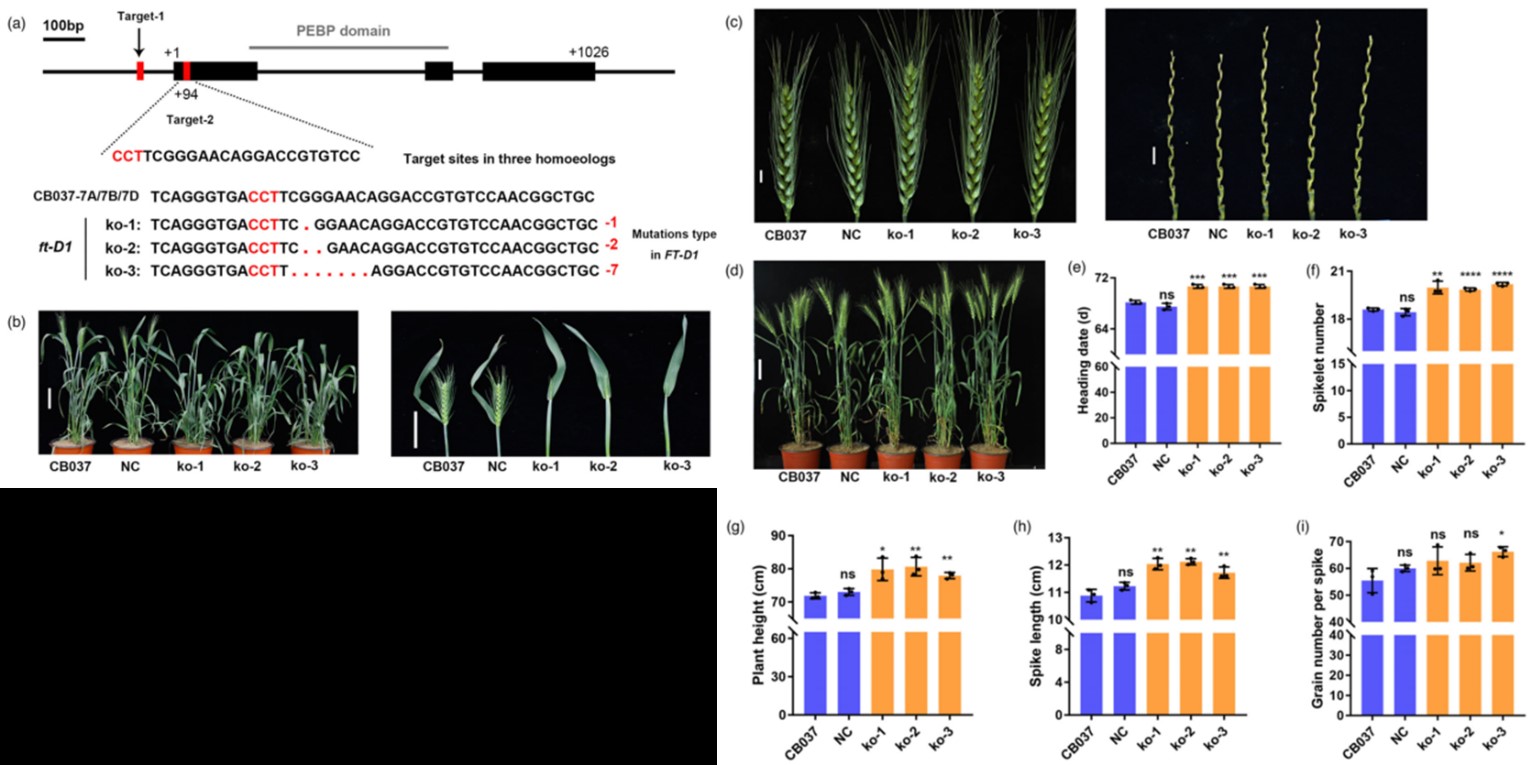
A single nucleotide deletion in the third exon of FT-D1 increases the spikelet number and delays heading date in wheat (Triticum aestivum L.)
The spikelet number and heading date are two crucial and correlated traits for yield in wheat. Here, a quantitative trait locus (QTL) analysis was conducted in F8 recombinant inbred lines (RILs) derived from crossing two common wheats with different spikelet numbers. A total of 15 stable QTL influencing total spikelet number (TSN) and heading date (HD) were detected. Notably, FT-D1, a well-known flowering time gene in wheat, was located within the finely mapped interval of a major QTL on 7DS (QTsn/Hd.cau-7D). A causal indel of one G in the third exon of FT-D1 was significantly associated with total spikelet number and heading date. Consistently, CRISPR/Cas9 mutant lines with homozygous mutations in FT-D1 displayed an increase in total spikelet number and heading date when compared with wild type. Moreover, one simple and robust marker developed according to the polymorphic site of FT-D1 revealed that this one G indel had been preferentially selected to adapt to different environments. Collectively, these data provide further insights into the genetic basis of spikelet number and heading date, and the diagnostic marker of FT-D1 will be useful for marker-assisted pyramiding in wheat breeding.(plant biotechnology journal)
QTL mapping for grain yield and three yield components in a population derived from two high‑yielding spring wheat cultivars
Genetic manipulation of yield components is an important approach to increase grain yield in wheat (Triticum aestivum). The present study used a mapping population comprised of 181 doubled haploid lines derived from two high-yielding spring wheat cultivars, UI Platinum and LCS Star. The two cultivars and the derived population were assessed for six traits in eight feld trials primarily in Idaho in the USA. The six traits were grain yield, fertile spikelet number per spike, productive tiller number per unit area, thousand kernel weight, heading date, and plant height. Quantitative Trait Locus (QTL) analysis of the six traits was conducted using 14,236 single-nucleotide polymorphism (SNP) markers generated from the wheat 90 K SNP and the exome and promoter capture arrays. Of the 19 QTL detected, 14 were clustered in four chromosomal regions on 4A, 6A, 7B and 7D. Each of the four QTL clusters was associated with multiple yield component traits, and these traits were often negatively correlated with one another. As a result, additional QTL dissection studies are needed to optimize trade-ofs among yield component traits for specifc production environments. Kompetitive allele-specifc PCR markers for the four QTL clusters were developed and assessed in an elite spring wheat panel of 170 lines, and eight of the 14 QTL were validated. The two parents contain complementary alleles for the four QTL clusters, suggesting the possibility of improving grain yield via genetic recombination of yield component loci.(Theoretical and Applied Genetics)
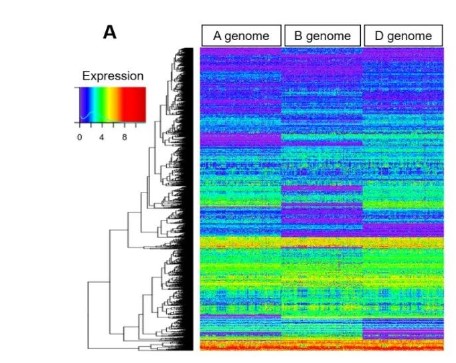
Genomic variants affecting homoeologous gene expression dosage contribute to agronomic trait variation in allopolyploid wheat
Allopolyploidy greatly expands the range of possible regulatory interactions among functionally redundant homoeologous genes. However, connection between the emerging regulatory complexity and expression and phenotypic diversity in polyploid crops remains elusive. Here, we use diverse wheat accessions to map expression quantitative trait loci (eQTL) and evaluate their effects on the population-scale variation in homoeolog expression dosage. The relative contribution of cis- and trans-eQTL to homoeolog expression variation is strongly affected by both selection and demographic events. Though trans-acting effects play major role in expression regulation, the expression dosage of homoeologs is largely influenced by cis-acting variants, which appear to be subjected to selection. The frequency and expression of homoeologous gene alleles showing strong expression dosage bias are predictive of variation in yield-related traits, and have likely been impacted by breeding for increased productivity. Our study highlights the importance of genomic variants affecting homoeolog expression dosage in shaping agronomic phenotypes and points at their potential utility for improving yield in polyploid crops.(Nature Communications)
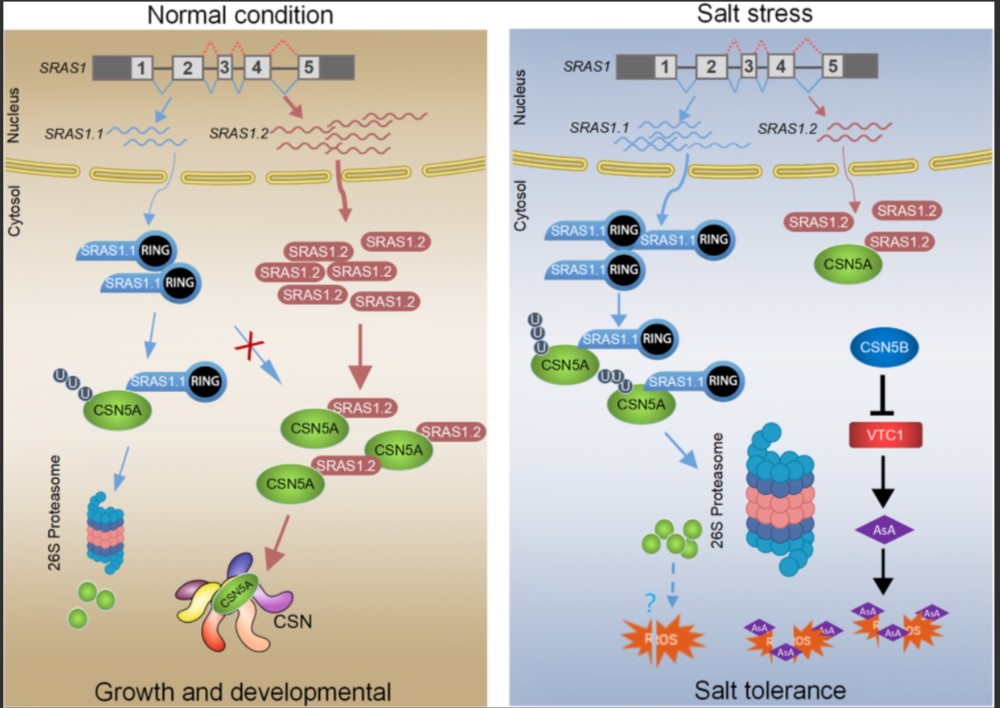
Salt responsive alternative splicing of a RING finger E3 ligase modulates the salt stress tolerance by fine-tuning the balance of COP9 signalosome subunit 5A
Increasing evidence points to the tight relationship between alternative splicing (AS) and the salt stress response in plants. However, the mechanisms linking these two phenomena remain unclear. In this study, we have found that Salt-Responsive Alternatively Spliced gene 1 (SRAS1), encoding a RING-Type E3 ligase, generates two splicing variants: SRAS1.1 and SRAS1.2, which exhibit opposing responses to salt stress. The salt stress-responsive AS event resulted in greater accumulation of SRAS1.1 and a lower level of SRAS1.2. Comprehensive phenotype analysis showed that overexpression of SRAS1.1 made the plants more tolerant to salt stress, whereas overexpression of SRAS1.2 made them more sensitive. In addition, we successfully identified the COP9 signalosome 5A (CSN5A) as the target of SRAS1. CSN5A is an essential player in the regulation of plant development and stress. The full-length SRAS1.1 promoted degradation of CSN5A by the 26S proteasome. By contrast, SRAS1.2 protected CSN5A by competing with SRAS1.1 on the same binding site. Thus, the salt stress-triggered AS controls the ratio of SRAS1.1/SRAS1.2 and switches on and off the degradation of CSN5A to balance the plant development and salt tolerance. Together, these results provide insights that salt-responsive AS acts as post-transcriptional regulation in mediating the function of E3 ligase.(SPLOS GENETICS)
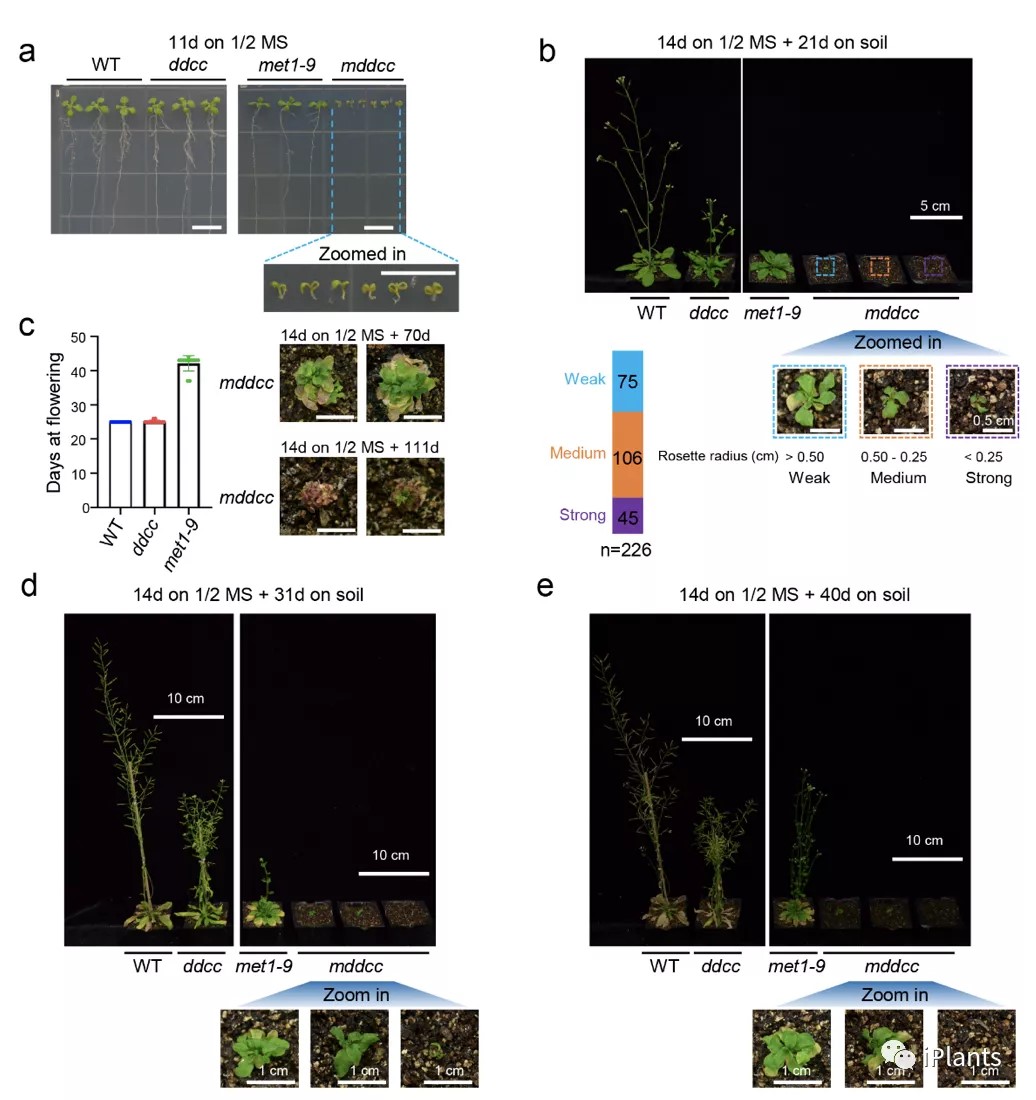
DNA methylation-free Arabidopsis reveals crucialroles of DNA methylation in regulating geneexpression and development
A contribution of DNA methylation to defense against invading nucleic acids and maintenance of genome integrity is uncontested; however, our understanding of the extent of involvement of this epigenetic mark in genome-wide gene regulation and plant developmental control is incomplete. Here, we knock out all five known DNA methyltransferases in Arabidopsis, generating DNA methylation-free plants. This quintuple mutant exhibits a suite of developmental defects, unequivocally demonstrating that DNA methylation is essential for multiple aspects of plant development. We show that CG methylation and non-CG methylation are required for a plethora of biological processes, including pavement cell shape, endoreduplication, cell death, flowering, trichome morphology, vasculature and meristem development, and root cell fate determination. Moreover, we find that DNA methylation has a strong dose-dependent effect on gene expression and repression of transposable elements. Taken together, our results demonstrate that DNA methylation is dispensable for Arabidopsis survival but essential for the proper regulation of multiple biological processes.(Nature Communications)
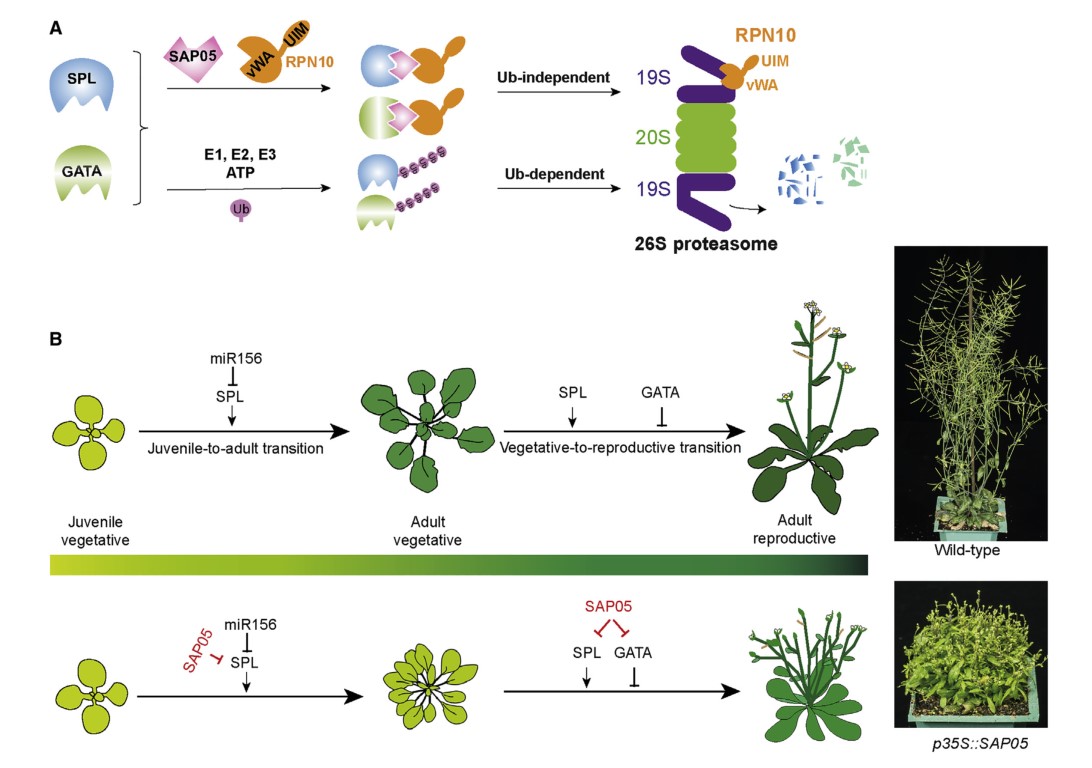
Parasitic modulation of host development by ubiquitin-independent protein degradation
Certain obligate parasites induce complex and substantial phenotypic changes in their hosts in ways that favor their transmission to other trophic levels. However, the mechanisms underlying these changes remain largely unknown. Here we demonstrate how SAP05 protein effectors from insect-vectored plant pathogenic phytoplasmas take control of several plant developmental processes. These effectors simultaneously prolong the host lifespan and induce witches’ broom-like proliferations of leaf and sterile shoots, organs colonized by phytoplasmas and vectors. SAP05 acts by mediating the concurrent degradation of SPL and GATA developmental regulators via a process that relies on hijacking the plant ubiquitin receptor RPN10 independent of substrate ubiquitination. RPN10 is highly conserved among eukaryotes, but SAP05 does not bind insect vector RPN10. A two-amino-acid substitution within plant RPN10 generates a functional variant that is resistant to SAP05 activities. Therefore, one effector protein enables obligate parasitic phytoplasmas to induce a plethora of developmental phenotypes in their hosts.(Cell)
Genetic analysis and gene mapping of a dwarf and liguleless mutation in barley
Leaf development underlies crop growth and productivity and has been a major target of crop domestication and improvement. However, most genes controlling leaf development in barley remain unknown.We identified a dwarf and liguleless (dl) mutant derived by ethylmethane sulfonate mutagenesis. The dl mutant showed dramatic changes in shoot architecture compared with wild-type (Yangnongpi 5) plants.Besides lacking ligules, the dl mutant showed much shorter plant height (28 cm) than Yangnongpi 5 (78 cm). By map-based cloning, the dl gene was localized to a 56.58-kb genomic interval on the long arm of chromosome 7. A C-to-T single-nucleotide substitution was identified at exon position 790, and is a functional mutation resulting in a proline-to-serine substitution at the 264th amino acid residue of HORVU7Hr1G106960. Consequently, HORVU7Hr1G106960 was identified as the DL gene, encoding 269 amino acids and containing the Arabidopsis LSH1 and Oryza G1 (ALOG) domain. DL is highly similar to rice OsG1-LIKE 1/2 (OsG1L1/2) and sorghum AWN1/AWN1-10 at the amino acid level. Although the dl mutant allele showed no expression changes in selected tissues by real-time PCR, we propose HORVU7Hr1G106960 as a candidate gene conferring the dwarf and liguleless phenotype in barley.(The Crop Journal)

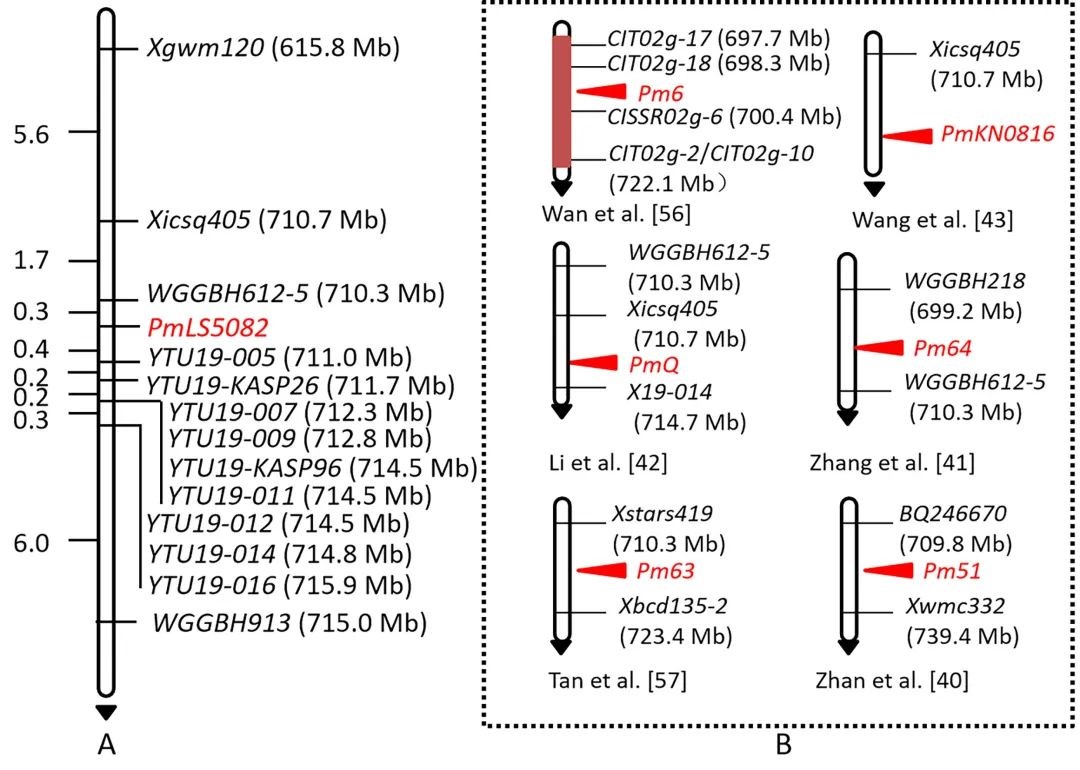
Genetic dissection of the powdery mildew resistance in wheat breeding line LS5082 using BSR-Seq
Powdery mildew of wheat is a destructive disease seriously threatening yield and quality worldwide. Comprehensive dissection of new resistance-related loci/genes is necessary to control this disease. LS5082 is a Chinese wheat breeding line with resistance to powdery mildew. Genetic analysis, using the populations of LS5082 and three susceptible parents (Shannong 29, Shimai 22 and Huixianhong), indicated that a single dominant gene, tentatively designated PmLS5082, conferred seedling resistance to different Blumeria graminis f. sp. tritici (Bgt) isolates. Bulked segregant RNA-Seq was carried out to map PmLS5082 and to profifile differentially expressed genes associated with PmLS5082. PmLS5082 was mapped to a 0.7 cM genetic interval on chromosome arm 2BL, which was aligned to a 0.7 Mb physical interval of 710.3–711.0 Mb. PmLS5082 differs from the known powdery mildew (Pm) resistance genes on chromosome arm 2BL based on their origin, chromosome positions and/or resistance spectrum, suggesting PmLS5082 is most likely a new Pm gene/allele. Through clusters of orthologous groups and kyoto encyclopedia of genes and genomes analyses, differentially expressed genes (DEGs) associated with PmLS5082 were profifiled. Six DEGs in the PmLS5082 interval were confifirmed to be associated with PmLS5082 via qPCR analysis, using an additional set of wheat samples and time-course analysis postinoculation with Bgt isolate E09. Ten closely linked markers, including two kompetitive allele-specifific PCR markers, were confifirmed to be suitable for marker-assisted selection of PmLS5082 in different genetic backgrounds, thus can be used to detect PmLS5082 and pyramid it with other genes in breeding programs.(The Crop Journal)