A hybrid inorganic–biological artificial photosynthesis system for energy-efficient food production
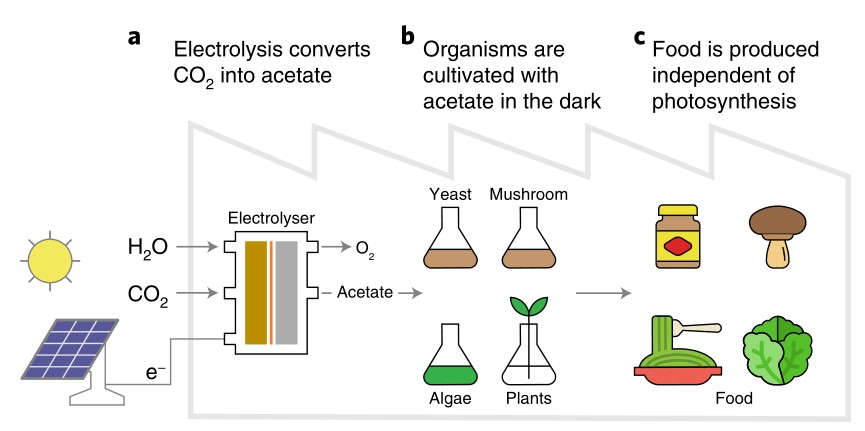
Artificial photosynthesis systems are proposed as an efficient alternative route to capture CO2 to produce additional food for growing global demand. Here a two-step CO2 electrolyser system was developed to produce a highly concentrated acetate stream with a 57% carbon selectivity (CO2 to acetate), allowing its direct use for the heterotrophic cultivation of yeast, mushroom-producing fungus and a photosynthetic green alga, in the dark without inputs from biological photosynthesis. An evaluation of nine crop plants found that carbon from exogenously supplied acetate incorporates into biomass through major metabolic pathways. Coupling this approach to existing photovoltaic systems could increase solar-to-food energy conversion efficiency by about fourfold over biological photosynthesis, reducing the solar footprint required. This technology allows for a reimagination of how food can be produced in controlled environments.(Nature Food)
Characterization of regulatory modules controlling leaf angle in maize
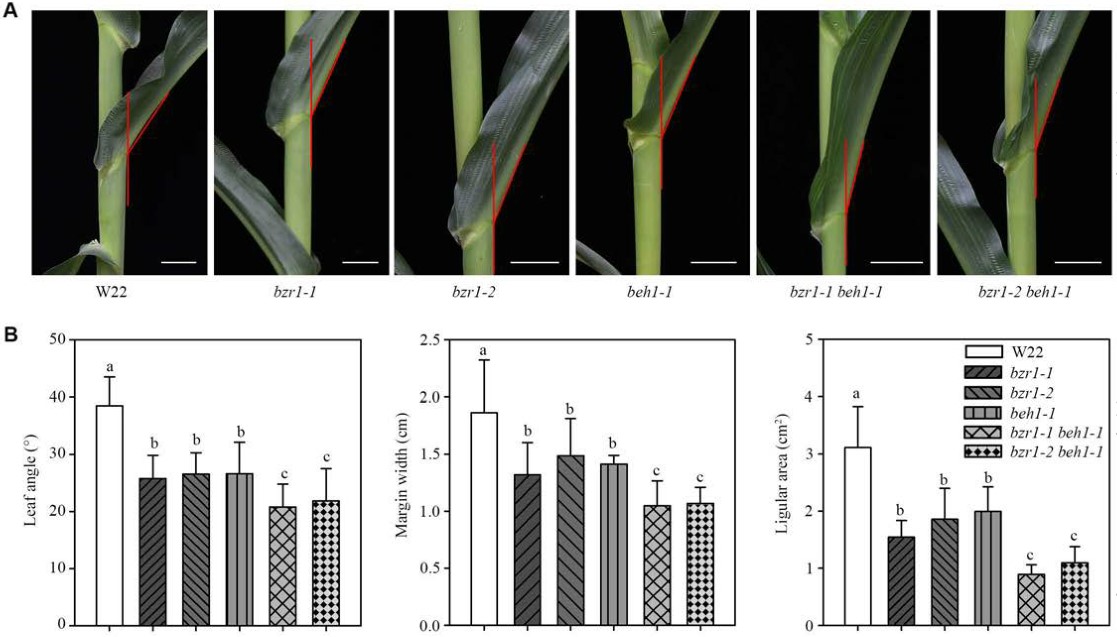
Leaf angle is an important agronomic trait determining maize (Zea mays) planting density and light penetration into the canopy and contributes to the yield gain in modern maize hybrids. However, little is known about the molecular mechanisms underlying leaf angle beyond the ZmLG1(liguleless1) and ZmLG2(Liguleless2 genes. In this study, we found that the transcription factor ZmBEH1 (BZR1/BES1 homolog gene 1) is targeted by ZmLG2 and regulates leaf angle formation by influencing sclerenchyma cell layers on the adaxial side. ZmBEH1 interacted with the transcription factor ZmBZR1 (Brassinazole Resistant 1), whose gene expression was also directly activated by ZmLG2. Both ZmBEH1 and ZmBZR1 bound to the promoter of ZmSCL28 (SCARECROW-LIKE 28), a third transcription factor that influences leaf angle. Our study demonstrates regulatory modules controlling leaf angle and provides gene editing targets for creating optimal maize architecture suitable for dense planting.(Plant Physiology)
Graph pangenome captures missing heritability and empowers tomato breeding
Missing heritability in genome-wide association studies defines a major problem in genetic analyses of complex biological traits1,2. The solution to this problem is to identify all causal genetic variants and to measure their individual contributions3,4. Here we report a graph pangenome of tomato constructed by precisely cataloguing more than 19 million variants from 838 genomes, including 32 new reference-level genome assemblies. This graph pangenome was used for genome-wide association study analyses and heritability estimation of 20,323 gene-expression and metabolite traits. The average estimated trait heritability is 0.41 compared with 0.33 when using the single linear reference genome. This 24% increase in estimated heritability is largely due to resolving incomplete linkage disequilibrium through the inclusion of additional causal structural variants identified using the graph pangenome. Moreover, by resolving allelic and locus heterogeneity, structural variants improve the power to identify genetic factors underlying agronomically important traits leading to, for example, the identification of two new genes potentially contributing to soluble solid content. The newly identified structural variants will facilitate genetic improvement of tomato through both marker-assisted selection and genomic selection. Our study advances the understanding of the heritability of complex traits and demonstrates the power of the graph pangenome in crop breeding.(Nature)
Exaggerated false positives by popular differential expression methods when analyzing human population samples
When identifying differentially expressed genes between two conditions using human population RNA-seq samples, we found a phenomenon by permutation analysis: two popular bioinformatics methods, DESeq2 and edgeR, have unexpectedly high false discovery rates. Expanding the analysis to limma-voom, NOISeq, dearseq, and Wilcoxon rank-sum test, we found that FDR control is often failed except for the Wilcoxon rank-sum test. Particularly, the actual FDRs of DESeq2 and edgeR sometimes exceed 20% when the target FDR is 5%. Based on these results, for population-level RNA-seq studies with large sample sizes, we recommend the Wilcoxon rank-sum test.(Genome Biology)
Genome-wide association study revealed TaHXK3-2A as a candidate gene controlling stomatal index in wheat seedlings
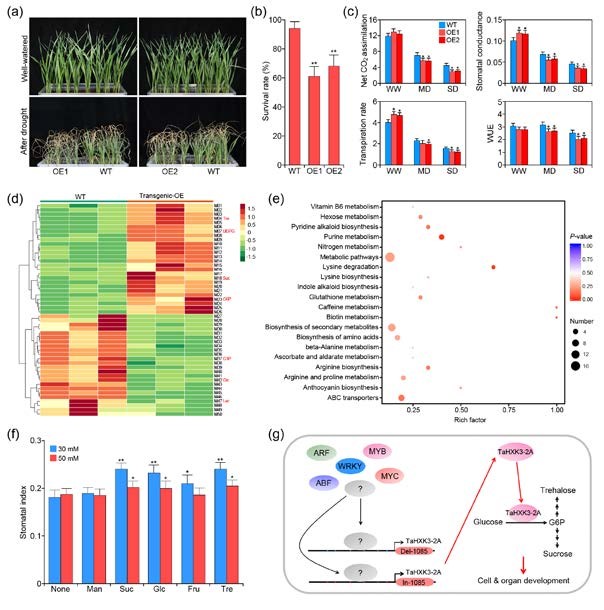
Stomata are important channels for the control of gas exchange between plants and the atmosphere. To examine the genetic architecture of wheat stomatal index, we performed a genome-wide association study (GWAS) using a panel of 539 wheat accessions and 450 678 polymorphic single nucleotide polymorphisms (SNPs) that were detected using wheat-specific 660K SNP array. A total of 130 SNPs were detected to be significantly associated with stomatal index in both leaf surfaces of wheat seedlings. These significant SNPs were distributed across 16 chromosomes and involved 2625 candidate genes which participate in stress response, metabolism and cell/organ development. Subsequent bulk segregant analysis (BSA), combined with GWAS identified one major haplotype on chromosome 2A, that is responsible for stomatal index on the abaxial leaf surface. Candidate gene association analysis revealed that genetic variation in the promoter region of the hexokinase gene TaHXK3-2A was significantly associated with the stomatal index. Moreover, transgenic analysis confirmed that TaHXK3-2A overexpression in wheat decreased the size of leaf pavement cells but increased stomatal density through the glucose metabolic pathway, resulting in drought sensitivity among TaHXK3-2A transgenic lines due to an increased transpiration rate. Taken together, these results provide valuable insights into the genetic control of the stomatal index in wheat seedlings.(Plant Cell Environ )
A telomere-to-telomere gap-free reference genome of watermelon and its mutation library provide important resources for gene discovery and breeding
Watermelon, Citrullus lanatus, is the world's third largest fruity crop. Reference genomes with gaps and narrow genetic base hinder functional genomics and genetic improvement of watermelon. Here, we report the assembly of a telomere-to-telomere (T2T) gap-free genome of the elite watermelon inbred G42 by incorporating the high-coverage and accurate long-read sequence data with multiple assembly strategies. All 11 chromosomes have been assembled into single contig pseudomolecules without gap, representing the highest completeness and assembly quality till now. The G42 reference genome contained a total length of 369,321,829 bp and 24,205 predicted protein-coding genes, with all 22 telomeres and 11 centromeres characterized. Over 200,000 M1 seeds from inbred G42 were generated using pollen EMS mutagenesis. In a sampling pool, 48 monogenic phenotypic mutations, selected from 223 M1 and 78 M2 mutants with morphological changes, were confirmed. The average density of mutation is 1 SNP/1.69 Mb and 1 indel/4.55 Mb per M1 plant, and 1 SNP/1.08 Mb and 1 indel/6.25 Mb per M2 plant. Taking advantage of the gap-free G42 genome, 8,039 mutations from the 32 plants sampled from M1 and M2 families were identified with 100% accuracy, whereas only 25% of the randomly selected mutations identified using 97103v2 reference genome could be confirmed. Using this library and the gap-free genome, two genes responsible for elongated fruit shape and male sterility (ClMS1) were identified, both being caused by a single base change from G to A. The validated gap-free genome and its EMS mutation library provide invaluable resources for functional genomics and genetic improvement of watermelon.(Mol Plant)

Biofortified tomatoes provide a new route to vitamin D sufficiency
Poor vitamin D status is a global health problem; insufficiency underpins higher risk of cancer, neurocognitive decline and all-cause mortality. Most foods contain little vitamin D and plants are very poor sources. We have engineered the accumulation of provitamin D3 in tomato by genome editing, modifying a duplicated section of phytosterol biosynthesis in Solanaceous plants, to provide a biofortified food with the added possibility of supplement production from waste material.(Nature Plant)
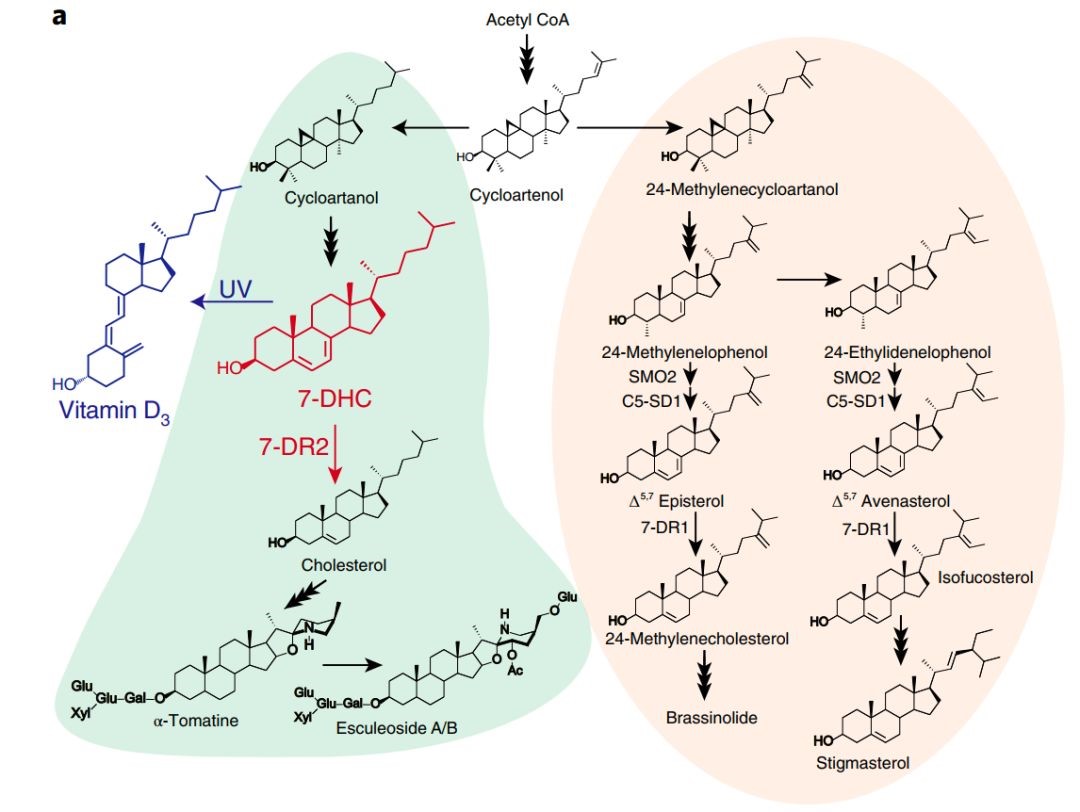
Advances in technology and manufacturing could boost the uptake of molecular farming
Therapeutic proteins such as vaccines, antibodies, hormones, and cytokines are generally produced in bacteria or eukaryotic systems, including chicken eggs and mammalian or insect cell cultures, with high production yield according to well-defined regulatory guidelines (1). The use of plants for the production of therapeutic proteins, called molecular farming, was proposed as an alternative biomanufacturing method in 1986. The first and only plant-derived therapeutic protein for human use was approved in 2012 for the treatment of Gaucher disease. In 2019, a plant-produced influenza virus vaccine completed phase 3 clinical trials, with encouraging results (2). More recently, phase 3 trials for an adjuvanted plant-made vaccine (CoVLP) against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (NCT04636697) began in March 2021. These successes have revived interest in plant-produced pharmaceuticals for human use, which could include edible drugs.(Science)
Genome-wide association study identifies novel candidate loci or genes affecting stalk strength in maize
Abstract: Stalk strength increases resistance to stalk lodging, which causes maize (Zea mays L.) production losses worldwide. The genetic mechanisms regulating stalk strength remain unclear. In this study, three stalk strength-related traits (rind penetrometer resistance, stalk crushing strength, and stalk bending strength) and four plant architecture traits (plant height, ear height, stem diameter, stem length) were measured in three field trials. Substantial phenotypic variation was detected for these traits. A genome-wide association study (GWAS) was conducted using general and mixed linear models and 372,331 single-nucleotide polymorphisms (SNPs). A total of 94 quantitative trait loci including 241 SNPs were detected. By combining the GWAS data with public gene expression data, 56 candidate genes within 50 kb of the significant SNPs were identified, including genes encoding flavonol synthase (GRMZM2G069298, ZmFLS2), nitrate reductase (GRMZM5G878558, ZmNR2), glucose-1-phosphate adenylyltransferase (GRMZM2G027955), and laccase (GRMZM2G447271). Resequencing GRMZM2G069298 and GRMZM5G878558 in all tested lines revealed respectively 47 and 2 variants associated with RPR. Comparison of the RPR of the zmnr2 EMS mutant and the wild-type plant under high- and low-nitrogen conditions verified the GRMZM5G878558 function. These findings may be useful for clarifying the genetic basis of stalk strength. The identified candidate genes and variants may be useful for the genetic improvement of maize lodging resistance.(The Crop Journal)
A hybrid inorganic–biological artificial photosynthesis system for energy-efficient food production
Artificial photosynthesis systems are proposed as an efficient alternative route to capture CO2 to produce additional food for growing global demand. Here a two-step CO2 electrolyser system was developed to produce a highly concentrated acetate stream with a 57% carbon selectivity (CO2 to acetate), allowing its direct use for the heterotrophic cultivation of yeast, mushroom-producing fungus and a photosynthetic green alga, in the dark without inputs from biological photosynthesis. An evaluation of nine crop plants found that carbon from exogenously supplied acetate incorporates into biomass through major metabolic pathways. Coupling this approach to existing photovoltaic systems could increase solar-to-food energy conversion efficiency by about fourfold over biological photosynthesis, reducing the solar footprint required. This technology allows for a reimagination of how food can be produced in controlled environments.(Nature Food)
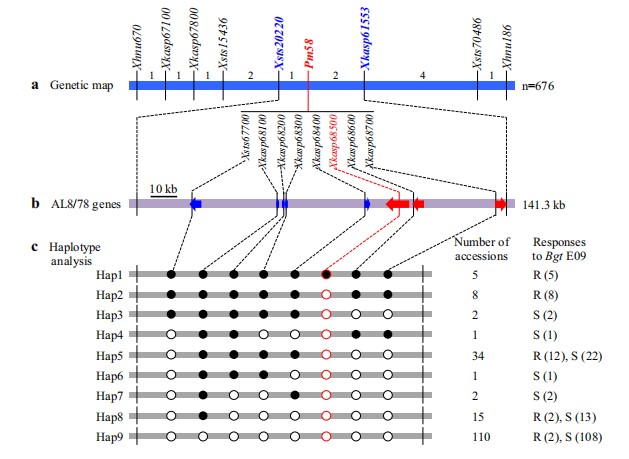
Fine mapping of Pm58 from Aegilops tauschii conferring powdery mildew resistance
Pm58 is a powdery mildew resistance gene identifed in Aegilops tauschii accession TA1662 and efective in a common wheat background. To finely map Pm58, an F2 population of 676 plants derived from the cross T093×TA1662 was used for recombinant screening. We obtained 13 recombinants that occurred between the fanking markers Xhnu670 and Xhnu186. Genotyping and phenotyping these recombinant F2:3 families delimited Pm58 to a 0.22-cM interval (Xsts20220– Xkasp61553) on chromosome arm 2DS. The region carrying the Pm58 locus was approximately 141.3-kb, which contained eight annotated genes according to the reference genome sequence of Ae. tauschii AL8/78. Haplotype analysis of 178 Ae. tauschii accessions using the candidate gene-specifc markers identifed a disease resistance gene AET2Gv20068500 as a candidate for Pm58. Comparative mapping of the Pm58-containing interval revealed two presence/absence variations (PAVs) between AL8/78 and common wheat Chinese Spring. PAV-1 resides in the 3′-end of AET2Gv20068500. The majority of 158 common wheat cultivars (84.8%) displayed the absence of a 14.1-kb fragment in the PAV-1 region, which was confrmed by aligning the targeted genome sequences of the other sequenced Ae. tauschii accessions and common wheat cultivars. A co-segregating marker Xkasp68500 developed from AET2Gv20068500 can distinguish TA1662 from all randomly selected common wheat cultivars and will be instrumental for tracking Pm58 in breeding programs.(Theoretical and Applied Genetics)
Enhanced stripe rust resistance obtained by combining Yr30 with a widely dispersed, consistent QTL on chromosome arm 4BL
Cultivars with durable resistance are the most popular means to control wheat stripe rust. Durable resistance can be achieved by stacking multiple adult plant resistance (APR) genes that individually have relatively small efect. Chinese wheat cultivars Ruihua 520 (RH520) and Fengdecun 12 (FDC12) confer partial APR to stripe rust across environments. One hundred and seventy recombinant inbred lines from the cross RH520×FDC12 were used to determine the genetic basis of resistance and identify genomic regions associated with stripe rust resistance. Genotyping was carried out using 55 K SNP array, and eight quantitative trait loci (QTL) were detected on chromosome arms 2AL, 2DS, 3BS, 4BL, 5BL (2), and 7BL (2) by inclusive composite interval mapping. Only QYr.nwafu-3BS from RH520 and QYr.nwafu-4BL.2 (named YrFDC12 for convenience) from FDC12 were consistent across the four testing environments. QYr.nwafu-3BS is likely the pleiotropic resistance gene Sr2/Yr30. YrFDC12 was mapped in a 2.1-cM interval corresponding to 12 Mb and fanked by SNP markers AX-111121224 and AX-89518393. Lines harboring both Yr30 and YrFDC12 displayed higher resistance than the parents and expressed pseudo-black chaf (PBC) controlled by loci Pbc1 and PbcFDC12, which co-segregated with Yr30 and YrFDC12, respectively. Both marker-based and pedigree-based kinship analyses revealed that YrFDC12 was inherited from founder parent Zhou 8425B. Fifty-four other wheat cultivars shared the YrFDC12 haplotype. These results suggest an efective pyramiding strategy to acquire highly efective, durable stripe rust resistance in breeding.(Theoretical and Applied Genetics)

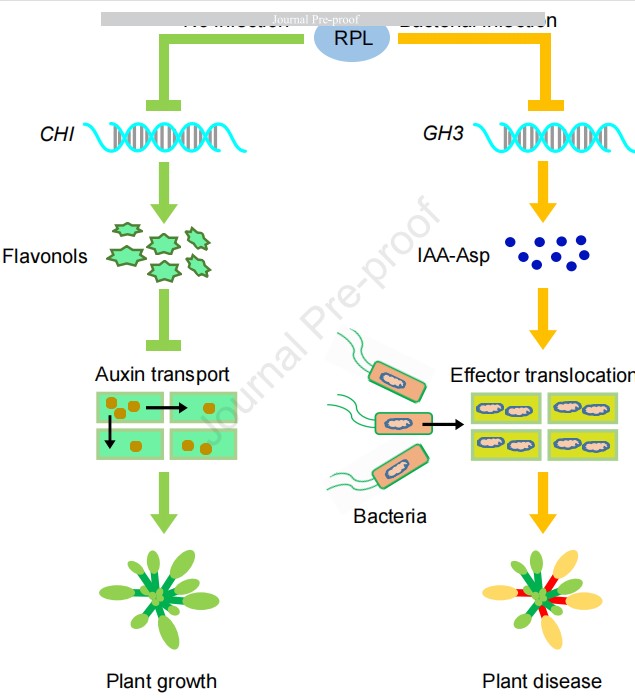
Natural variation in the transcription factor Replumless contributes to both disease resistance and plant growth in Arabidopsis
When attacked by pathogens, plants need to reallocate energy from growth to defense to fend off the invaders, which frequently incurs growth penalties. This phenomenon is known as growth-defense tradeoff, which is orchestrated by a hardwired transcriptional network. Altering key factors involved in this network has the potential to increase disease resistance without growth or yield loss, but the mechanisms underlying such changes need further investigation. By conducting a genome-wide association study (GWAS) of leaves infected by hemi-biotrophic bacterial pathogen Pseudomonas syringae pv. tomato (Pst) DC3000, we discovered that the Arabidopsis transcription factor REPLUMLESS (RPL) is necessary for bacterial resistance. More importantly, RPL functions in promoting both disease resistance and growth. Transcriptome analysis revealed a cluster of genes in the GRETCHEN HAGEN 3 (GH3) family that were significantly up-regulated in the rpl mutants, which led to the accumulation of indole-3-acetic acid-aspartic acid (IAA-Asp). Consistent with this observation, transcripts of virulence effector genes were activated by IAA-Asp accumulated in the rpl mutants. We found that RPL protein could directly bind to GH3 promoters and repress their expression. On the other hand, RPL repressed flavonol synthesis through directly repressing CHI expression and thus activated the auxin transport pathway, which promotes plant growth. Therefore, RPL plays an important role in plant immunity and functions in the auxin pathway to optimize Arabidopsis growth and defense.(Plant Communications)
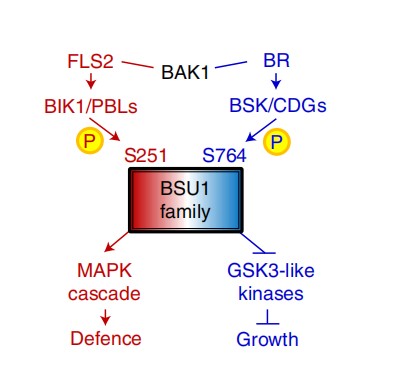
Deconvoluting signals downstream of growth and immune receptor kinases by phosphocodes of the BSU1 family phosphatases
Hundreds of leucine-rich repeat receptor kinases (LRR-RKs) have evolved to control diverse processes of growth, development and immunity in plants, but the mechanisms that link LRR-RKs to distinct cellular responses are not understood. Here we show that two LRR-RKs, the brassinosteroid hormone receptor BRASSINOSTEROID INSENSITIVE 1 (BRI1) and the flagellin receptor FLAGELLIN SENSING 2 (FLS2), regulate downstream glycogen synthase kinase 3 (GSK3) and mitogen-activated protein (MAP) kinases, respectively, through phosphocoding of the BRI1-SUPPRESSOR1 (BSU1) phosphatase. BSU1 was previously identified as a component that inactivates GSK3s in the BRI1 pathway. We surprisingly found that the loss of the BSU1 family phosphatases activates effector-triggered immunity and impairs flagellin-triggered MAP kinase activation and immunity. The flagellin-activated BOTRYTIS-INDUCED KINASE 1 (BIK1) phosphorylates BSU1 at serine 251. Mutation of serine 251 reduces BSU1’s ability to mediate flagellin-induced MAP kinase activation and immunity, but not its abilities to suppress effector-triggered immunity and interact with GSK3, which is enhanced through the phosphorylation of BSU1 at serine 764 upon brassinosteroid signalling. These results demonstrate that BSU1 plays an essential role in immunity and transduces brassinosteroid–BRI1 and flagellin–FLS2 signals using different phosphorylation sites. Our study illustrates that phosphocoding in shared downstream components provides signalling specificities for diverse plant receptor kinases.(nature plants)
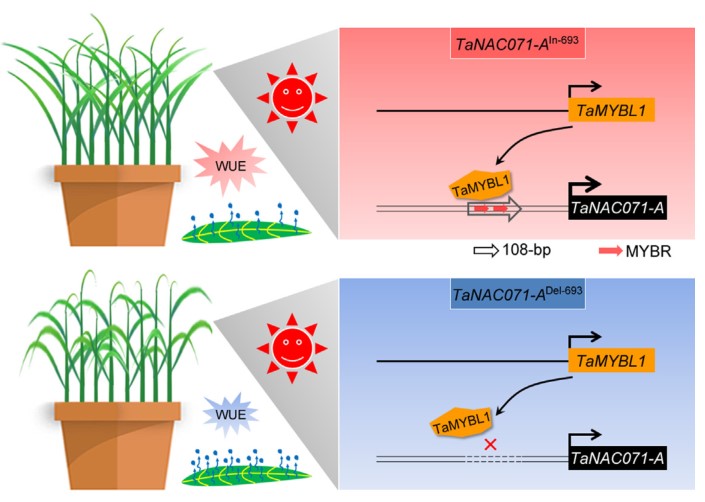
Variation in cis-regulation of a NAC transcription factor contributes to drought tolerance in wheat
Drought is a major environmental factor limiting wheat production worldwide, and developing drought-tolerant cultivars is a central challenge for wheat breeders globally. Therefore, it is important to identify genetic components determining drought tolerance in wheat. In this study, we identified a wheat NAC gene (TaNAC071-A) that is tightly associated with drought tolerance by a genome-wide association study. Knockdown of TaNAC071-A in wheat attenuated plant drought tolerance, whereas its overexpression significantly enhanced drought tolerance through improved water-use efficiency and increased expression of stress-responsive genes. This heightened water-saving mechanism mitigated the yield loss caused by water deficit. Further candidate gene association analysis showed that a 108-bp insertion in the promoter of TaNAC071-A alters its expression level and contributes to variation in drought tolerance among wheat accessions. This insertion contains two MYB cis-regulatory elements (CREs) that can be directly bound by the MYB transcription activator, TaMYBL1, thereby leading to increased TaNAC071-A expression and plant drought tolerance. Importantly, introgression of this 108-bp insertion allele, TaNAC071-A In-693, into drought-sensitive cultivars could improve their drought tolerance, demonstrating that it is a valuable genetic resource for wheat breeding. Taken together, our findings highlight a major breakthrough in determining the genetic basis underlying phenotypic variation in wheat drought tolerance and showcase the potential of exploiting CRE-containing indels for improving important agronomical traits.(Molecular Plant)
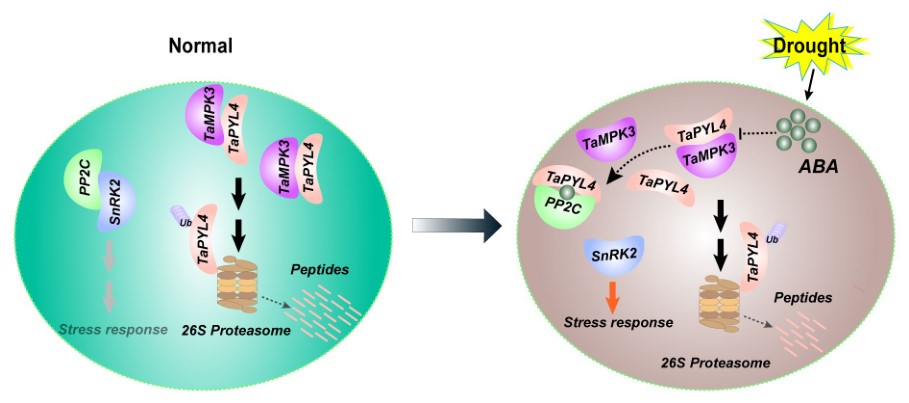
Mitogen-activated protein kinase TaMPK3 suppresses ABA response by destabilizing TaPYL4 receptor in wheat
Abscisic acid (ABA) receptors are considered as the targeted manipulation of ABA sensitivity and water productivity in plants. Regulation of their stability or activity will directly affect ABA signaling. Mitogen-activated protein kinase (MAPK) cascades link multiple environmental and plant developmental cues. However, the molecular mechanism of ABA signaling and MAPK cascade interaction remains largely elusive. TaMPK3 overexpression decreases drought tolerance and wheat sensitivity to ABA, significantly weakening ABA’s inhibitory effects on growth. Under drought stress, overexpression lines show lower survival rates, shoot fresh weight, and proline content, but higher malondialdehyde (MDA) levels at seedling stage, as well as decreased grain width and thousand grain weight in both greenhouse and field conditions at the adult stage. TaMPK3-RNAi increases drought tolerance. TaMPK3 interaction with TaPYL4 leads to decreased TaPYL4 levels by promoting its ubiquitin-mediated degradation, while ABA treatment diminishes TaMPK3-TaPYL interactions. In addition, the expression of ABA signaling proteins is impaired in TaMPK3-overexpressing wheat plants under ABA treatment. The MPK3-PYL interaction module was found to be conserved across monocots and dicots. Our results suggest that the MPK3-PYL module could serve as a negative regulatory mechanism for balancing appropriate drought stress response with normal plant growth signaling in wheat.(New phytologist)